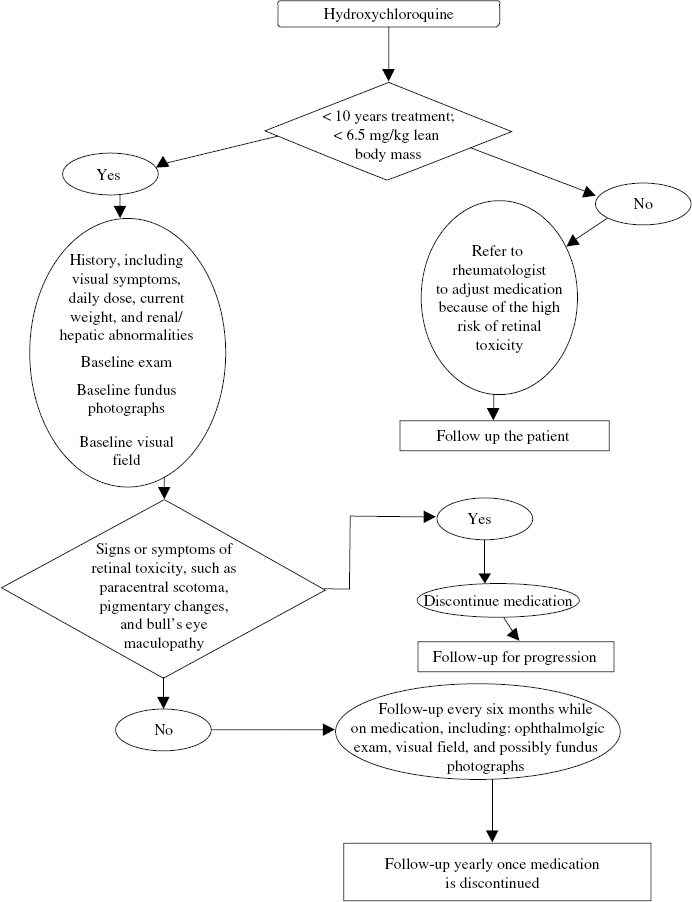
21 Diabetic retinopathy is the leading cause of blindness in the United States for people between the ages of 25 and 55 years. It is estimated that there are 16 million people in the United States who have diabetes.1 The incidence of diabetic retinopathy is 29% in people who have had diabetes for fewer than 5 years and up to 78% in those who have had the disease for 15 years or longer.2 Screening includes the education of patients as to the role of medical control in the prevention of sequelae as well as identifying patients who need treatment (see Chapter 23, Fig. 23–7). The use of laser photocoagulation for macular edema3 and proliferative disease,4 combined with current vitreoretinal surgical techniques,5,6 can reduce the risk of severe vision loss.1 Patients who develop diabetes during pregnancy, or gestational diabetes, do not have a significant risk of developing diabetic retinopathy during gestation.7 Gestational diabetes mellitus, however, is a major risk factor for the subsequent development of non–insulin-dependent diabetes with its associated complications. Patients with insulin-dependent diabetes mellitus (IDDM), or type 1 diabetes mellitus, have a low risk of retinopathy for the first 5 years following diagnosis.8 Thereafter, the risk increases. After 5 years, 25% of patients with IDDM have retinopathy. After 10 years, more than 50% have retinopathy, and after 15 years, about 80% have retinopathy. More than 30% will develop retinopathy that requires treatment.8 In adult-onset, non–insulin-dependent, or type II, diabetes mellitus, the risk is more difficult to specify because of the variability as to when in the course of disease the individual diagnosis is made. Patients may present with advanced retinopathy at diagnosis. Of type II patients with diabetes for fewer than 5 years, 24 to 40% have retinopathy, and after more than 15 years, 53 to 84% have retinopathy. About 2% of patients with type II diabetes will develop proliferative retinopathy within 5 years of being diagnosed, and 25% of patients will develop proliferative disease after 25 years.9 In addition to type and duration of diabetes, the incidence of diabetic retinopathy is dependent on several specific factors. The most important factors are duration of diabetes, level of glycemic control,2,10,11 control of serum cholesterol,12 and systemic hypertension. Each of these is an independent risk factor for the onset and progression of diabetic retinopathy. Smoking also adversely affects retinopathy.1 Duration of diabetes has long been known to be directly related to the complications. The results of the Diabetes Control and Complications Trial in 19912,10,13 proved the role of medical intervention with monitoring and control of serum glucose, blood pressure, and cholesterol. The level of hyperglycemia over time is directly related to the serum HbA1c level. Improving this value from the 9 range to the 7 range can reduce the onset and progression of retinopathy up to 75%.10 Significant but slightly lower reductions in nephropathy also were found. It was also proven that patients with lowered levels of serum cholesterol and treatment of elevated systemic blood pressure had significantly less severe retinopathy.13 Patients receiving interferon-beta for hepatitis or leukemia have been reported to be at risk for more rapid progression of retinopathy.14 The pathophysiology of diabetic retinopathy includes hyperglycemia-induced glycosylation of proteins that results in microvascular injury. Hemodynamic changes result from morphologic reticulocyte and leukocyte changes, hyperviscosity, as well as other factors. The injury of the microvasculature, ischemia, and subsequent release of factors cause vascular permeability and angiogenesis. An important growth factor is vascular endothelial growth factor (VEGF), which is thought to be important in the pathogenesis of both macular edema and preretinal neovascularization, both causing loss of vision.15 Nonproliferative diabetic retinopathy (NPDR) is characterized by clinical findings indicative of early injury to the microvasculature. Microaneurysms, intraretinal hemorrhages, cotton-wool spots, venous beading, and intraretinal microvascular abnormalities (IRMA) have been correlated to the patient’s risk of progression to proliferative disease.16 Progressive leakage secondary to compromised barrier function of retinal endothelial cells is believed to cause macular edema. Consequently, retinal thickening and hard exudates can involve or threaten the fovea. Clinically significant macular edema (CSME), defined as retinal thickening within 500 of the fovea or hard exudates associated with retinal thickening within 500 μ of the fovea or a disc area of thickening one disc diameter (DD) from the fovea (see Chapter 23, Table 23–2), is identified by biomicroscopic examination of the macula. Diagnosis often requires stereoscopic examination at the slit lamp with a contact lens. The role of focal or grid macular laser photocoagulation has been defined by the Early Treatment Diabetic Retinopathy Study (ETDRS). Treatment to leaking microaneurysms or areas of capillary leakage or nonperfusion identified by fluorescein angiography is recommended once CSME is diagnosed.3 The study showed 14% of laser-treated eyes versus 33% of untreated eyes had visual loss at 3 years. The use of vitreoretinal surgery and steroids delivered by different methods (for example, subtenon or intravitreal) in the treatment of CSME is also being evaluated. Greater evidence that inflammation may play a role in diabetes mellitus is being discovered through laboratory research.17 Attention is directed to the optic disc to determine whether neovascularization is present and, if so, its size. The macula is examined for neovascularization or macular thickening and exudation. The retinal area covered by the seven standard 30-degree images17 is important in most screening of diabetic retinopathy. Either biomicroscopic examination or review of the stereoscopic images is performed. To grade the level of NPDR, particular attention is given to quadrants involving the nasal and temporal fields around the optic nerve. Intraretinal hemorrhage, venous beading, and IRMA are identified. Severe NPDR is diagnosed with four quadrants of intraretinal hemorrhages, two quadrants of venous beading, or one quadrant of IRMA, indicating the risk of developing high-risk proliferative disease to be 15% within a year. For patients with very severe NPDR, defined by having any two of the above characteristics, the risk of developing high-risk proliferative disease is 45% within the year.3 Proliferative diabetic retinopathy (PDR) is defined by neovascularization of the disc (NVD) or neovascularization elsewhere (NVE). NVD or NVE can cause vitreous hemorrhage, retinal detachment, and, if untreated, blindness secondary to retinal detachment or neovascular glaucoma within 2 years in up to 40% of patients.4,18,19 Panretinal laser photocoagulation is recommended when a level of severity of neovascularization occurs (NVD of about 1:3 DD or greater or NVE of 1:2 DD or greater associated with vitreous or preretinal hemorrhage). Laser reduces the risk of severe vision loss by 50% or more.18–20 Diabetic patients may be asymptomatic, even when retinopathy is present. Patients may wait until significant visual symptoms develop before seeking ophthalmic care. Therefore, it is important that diabetic patients are educated as to the need for them to have timely ophthalmologic examinations. The American Academy of Ophthalmology (AAO) and American Diabetic Association (ADA) recommend yearly eye examinations for all diabetics unless more frequent examinations are recommended by the ophthalmologist. With macular edema, the patient may notice blurriness or distortion of vision. The development is often gradual and difficult for the patient to identify. The onset of floaters may indicate vitreous hemorrhage from proliferative retinopathy. Abrupt loss of vision may reflect vitreous hemorrhage, retinal detachment involving the macula, or sometimes diabetic papillitis. An ophthalmologist trained in the examination and treatment of diabetic retinopathy should be consulted for examination of the patient at risk. A history should be taken, noting the regimen of glycemic monitoring, use of hypoglycemic agents, glycosylated hemoglobin levels, blood pressure treatment, and cholesterol control.1 The timing of the first examination depends on the type of diabetes. Insulin-dependent diabetics usually have an accurate date of onset of their diabetic state. As it is rare to have significant complications from type I diabetes within the first 5 years of onset, the first ophthalmic examination may be scheduled at 5 years. In contrast, patients with non-IDDM may have had poor glycemic control for an unknown duration prior to diagnosis and should have the first examination closer to the time of diagnosis of diabetes. The frequency of ophthalmic examinations is determined by the level of severity of retinopathy on the initial examination and on the patient’s general health. Patients who are well controlled and expected to maintain appropriate follow-up care can be seen less frequently. Any patient who is poorly controlled, has a record of poor follow-up care, or is recently under stringent control may be seen more frequently. In the interim, the patient should notify the ophthalmologist with any major change in vision, such as reduced vision, distortion, or floaters. If no retinopathy is found during the initial examination, a follow-up examination may be conducted in 1 year. If there is mild to moderate NPDR present, the patient should be seen in the next 3 to 9 months. The potential for progression to CSME and proliferative changes should be considered.2 Patients with mild retinopathy may be seen for follow-up in 9 to 12 months. Patients with moderate retinopathy should be reevaluated in 4 to 6 months as the risk of developing CSME is 23% within 4 years.3 Patients with severe NPDR should be examined every 4 months and considered for laser therapy if there is concern that follow-up with examinations will be poor. The role of laser and its risks and benefits should be reviewed. Patients who develop macular edema should be checked every 2 to 4 months. If a patient develops CSME, he or she should be treated with focal or grid laser and followed every 6 to 12 weeks until edema is resolved.20 If resolution is not present at 4 months, retreatment is recommended. Patients with early PDR should be examined every 1 to 2 months until the development of high-risk characteristics, such as NVD and NVE. When NVD is 1:3 DD or greater or 1:2 DD of NVE with vitreous hemorrhage occurs, the patient should receive panretinal photocoagulation with close observation until the proliferation has regressed and become quiescent. In questionable cases, such as vitreous hemorrhage obscuring any NVE, an ultrasound is considered to rule out a retinal detachment and laser may be attempted, especially if the fellow eye demonstrates diabetic retinopathy. Patients with vitreous hemorrhage should be followed at least monthly to attempt panretinal photocoagulation and to check for retinal detachment. Patients are instructed to sleep with several pillows to permit settling of the vitreous hemorrhage inferiorly. Once quiescent and stable, examinations every 6 months are recommended.4 Patients who develop retinal detachment from fibrovascular membranes causing traction on the retina should be evaluated for possible vitreoretinal surgery. Guidelines regarding screening and follow-up of diabetic retinopathy during pregnancy are presented in Figure 21–1. Patients with gestational diabetes are not at risk for developing retinopathy7 and therefore do not need to be screened during pregnancy. Diabetic women with no history of diabetic retinopathy who become pregnant should have an eye examination during the first trimester. If no retinopathy is present, annual examinations will suffice unless visual symptoms occur.7 Forty-seven to 65% of women with preexisting NPDR will show progression of their retinopathy during pregnancy,7,21 and 5% will progress to proliferative disease at some point during their pregnancy.7 Therefore, it is necessary to follow pregnant diabetic patients more closely. Patients with mild to moderate NPDR should be examined during the first trimester and again during the third trimester. Women who develop or have preexisting PDR need to be followed closely because changes can progress rapidly. If no high-risk characteristics are present, examinations should be performed every month. If high-risk characteristics develop, panretinal photocoagulation should be performed; the patient should then be followed up frequently until the proliferation is quiescent. It should also be noted that proliferative changes in pregnancy can spontaneously regress in the late third trimester and early postpartum period.22 Patients who develop CSME should be considered for focal laser treatment and followed in 6 to 8 weeks. The macular edema, especially a diffuse type, may improve spontaneously after delivery.22 The risk of visual impairment and blindness from diabetic retinopathy increases directly related to control and duration of diabetes. The leading cause of blindness from diabetic retinopathy is macular edema. If visual acuity drops to 20/60 or lower, the patient is likely to have permanent vision loss despite laser treatment. In the absence of treatment or poor response to treatment, vision can continue to decline. Untreated PDR has a risk of progression to blindness within 2 years in 20 to 40% of cases.4 The HIV virus weakens the immune system and pre-disposes the eye to a number of opportunistic infections. The most common and visually devastating of these is cytomegalovirus (CMV) retinitis. Screening of the HIV infected patient is primarily directed at identifying CMV retinitis but includes the identification of other diseases that affect the retina and choroid. These include acute retinal necrosis (ARN), progressive outer retinal necrosis (PORN), ocular toxoplasmosis, Candida retinitis, Pneumocystis choroidopathy, cryptococcal choroiditis, syphilitic chorioretinitis, and mycobacterial infections.23,24 Human immunodeficiency virus itself can cause retinopathy. Cotton-wool spots are the most common manifestation of HIV retinopathy, but hemorrhages, microaneurysms, capillary nonperfusion, and telangiectatic vessels also occur.24,25 The retinopathy becomes more prevalent as the systemic disease progresses. HIV retinopathy is found in only about 1 to 3% of patients with asymptomatic HIV infection,24 whereas more than half of patients with acquired immunodeficiency syndrome (AIDS) have some signs of retinopathy. HIV retinopathy itself has minimal, if any, effects on vision24 but sometimes causes small scotomata or blind spots. It does need to be differentiated from the early stages of CMV retinitis because of the sometimes similar appearance of cotton-wool spots and early CMV retinitis. Patients who are severely immunosuppressed from HIV, chemotherapy for cancer, or iatrogenic immunosuppression for transplantation are at risk for developing CMV retinitis. Among these, patients with HIV and low CD4 counts are at the greatest risk for developing CMV retinitis. Typically, when it presents in the posterior pole, CMV retinitis appears as a yellowish white opacification of the retina, sometimes with hemorrhages and exudates. CMV retinitis in the peripheral retina can appear more granular with less hemorrhage. In either case, there is usually little inflammation within the vitreous. Early CMV retinitis can look like cotton-wool spots, but close follow-up with fundus photographs will reveal progressive increase in lesion size associated with CMV retinitis versus the eventual resolution of cotton-wool spots. A patient with active CMV retinitis may notice decreased vision, flashes, floaters, scotomata, or shadows. The patient should seek care immediately if any of these symptoms occur; however, patients may be asymptomatic and harbor CMV retinitis. Therefore, screening asymptomatic patients is considered when the CD4 count is low. The CD4 count is the most reliable gauge in determining when patients should be screened and scheduled for follow-up appointments. CMV retinitis rarely occurs in patients with CD4 counts greater than 100. CMV retinitis usually occurs when the count drops below 100, and most cases develop when the CD4 count is less than 50.25,26 It has been recommended that patients with CD4 counts less than 50 should have an eye examination every 3 to 4 months25 or immediately if any changes in vision occur. Although there are no specific guidelines for screening patients with CD4 counts greater than 50, patients with CD4 counts in the 50 to 100 range probably should be examined every 6 months because CD4 counts can go down and CMV retinitis can occur with CD4 counts over 50. Patients with CD4 counts greater than 100 should have at least yearly eye examinations. Although examinations are scheduled for CMV retinitis screening purposes, other HIV related retinal diseases should be considered in the differential diagnosis. An initial workup and treatment plan should be implemented if any signs of infection develop. Consultation with the patient’s infectious disease physician is necessary to assess for systemic infection and decide on appropriate therapy and workup. Once CMV retinitis is diagnosed, the patient should be treated with antivirals such as ganciclovir or foscarnet to reduce vision loss. Ganciclovir can be given intravenously or as an intravitreal insert. There is also an oral formulation available. Follow-up examinations every 1 to 2 weeks for the first month after intravenous induction therapy is initiated, and then monthly examinations thereafter are recommended.27,28 Even after successful treatment of CMV retinitis, patients must be followed up closely because relapse is common,28 and retinal detachments occur in about 25% of patients within months of developing the disease.29,30 In general, greater and more peripheral retinal involvement creates a greater likelihood of developing a retinal detachment.29,30 Active CMV retinitis also is associated more often with retinal detachments. The use of combination therapy is often needed in cases that progress. With the advent of highly active antiretroviral therapy (HAART), the prevalence of CMV retinitis is lessening. HAART enables the CD4 count to rise and the immune system to recover to some degree. Patients with a history of CMV retinitis who start HAART and have an increase in CD4 count and immune function can develop a condition called immune recovery vitreitis or immune reconstitution uveitis. In this condition, the CMV retinitis is inactive, but with improved immune function, an inflammatory response to CMV can occur. The larger the area of retina previously involved with CMV retinitis, the greater the likelihood of developing an inflammatory response. If the vitreitis is significant, decreased vision may result. Visual loss may occur secondary to proliferative vitreoretinopathy, cystoid macular edema, epiretinal membrane formation, or posterior subcapsular cataract.31,32 Given this, patients with inactive CMV retinitis and CD4 counts above 100 should be examined regularly to evaluate for uveitis as well as retinal tears and detachments. The patient should be told to contact his or her ophthalmologist immediately if any symptoms of uveitis (reduced vision, floaters) or retinal tears and detachment (flashes, floaters, visual field defect, reduced vision) occur between scheduled visits. Retinopathy of prematurity (ROP) initially was described as retrolental fibroplasia for the most severe stage of ROP, stage 5. The International Classification of Retinopathy of Prematurity (ICROP) recognized earlier stages in the pathogenesis, and the term ROP was adopted.33 Each year, ROP causes blindness to at least 550 infants and some loss of vision to thousands of others.34 In about 90% of the early stages 1 and 2, ROP may resolve spontaneously, but in the incompletely vascularized infant retina, ROP can develop, showing characteristic stages. The advanced stages lead to blindness. Screening is crucial in early diagnosis and in detecting threshold disease when the risk of vision loss approaches 50%.35 With treatment of threshold disease, reduced vision can be prevented in almost 50% of threshold cases.36 The Committee for the Classification of Retinopathy of Prematurity (CRYO-ROP) study published an international classification of ROP.33 This classification uses four parameters: (1) zone, (2) clock hours of involvement (circumferential extent of ROP), (3) stage of disease, and (4) vascular tortuosity and engorgement of retinal vessels (plus disease). The zone indicates the amount of mature or vascularized retina. Zone 1 is the most posterior zone, demarcated by a circle centered on the optic disc, the radius of which extends twice the distance from the disc to the fovea. Zone 2 is a circle centered on the optic disc with the radius corresponding to the distance from the optic disc to the nasal ora serrata. Zone 3 is the temporal part of peripheral retina not included in zones 1 and 2. Zone 3 ROP is often associated with favorable outcome and rarely requires treatment.31 The stage of ROP represents the level of progression of the disease. Stages 1 and 2 have an excellent prognosis and are monitored for progression or resolution. Stage 3 ROP has neovascularization between the avascular and vascularized retina. When threshold disease (see Chapter 20, Table 20–3) occurs, laser or cryotherapy is recommended. Stages 4 and 5 ROP are partial and total retinal detachment. The clock hours of involvement indicate the circumferential extent of the worst stage in an eye. The presence of plus disease is indicated in each of the posterior quadrants of an eye.37 Risk factors in the neonatal and antenatal period have been recognized as being associated with the progression of ROP.33,38 The most important criteria are birth weight and gestational age.33,38 The likelihood of occurrence of ROP and the severity of disease dramatically increase, the lower the birth weight. Among all risk factors, birth weight is the most significant predictive factor. In infants weighing between 1000 and 1250 g, about half will show some signs of ROP, and only 2% will reach threshold.33,38 According to Palmer, 65.8% of infants weighing less than 1251 g developed ROP, whereas 81.6% infants weighing less than 1000g developed the disease.38 The incidence of severe ROP is small in infants weighing more than 1500 g; however, some cases of late-stage ROP have been described in infants with birth weights exceeding the usual criteria. Gestational age (GA) is another risk factor. Infants with a GA of less than 27 weeks have been reported to have an ROP incidence of 83.4%. As many as 80% of these infants are diagnosed as having prethreshold ROP and 13% with threshold at less than 33 weeks corrected age (gestational age + chronological age); thus, it is recommended that the initial examination be scheduled within 4 to 6 weeks of neonatal life.35,39 It has been noted that racial predisposition exists: Caucasians appear more susceptible to developing ROP.38 High, uncontrolled oxygen is an important risk factor, recognized in the 1950s, especially if inspired oxygen concentration over 70 to 80% is used. Efforts to control oxygen reduced the incidence of ROP. Resurgence has occurred as methods to save lower-birth-weight infants were developed. The Supplemental Therapeutic Oxygen for Pre-threshold Retinopathy of Prematurity (STOP-ROP) study reported that supplemental therapeutic oxygen to cause a pulse oximetry saturation of 96 to 99% given at prethreshold disease39 did not cause progression to threshold ROP, nor did it reduce the number of infants requiring ablative surgery. A subgroup of infants with prethreshold ROP without plus disease showed benefit from supplemental oxygen but this was not statistically significant in the study design, and infants on supplemental oxygen had increased pulmonary morbidity.37,40 Other factors that might increase the risk of ROP include repeated blood transfusions, prolonged parental nutrition, infections, and respiratory distress. Maternal factors include multiple gestation, poor prenatal care, and drug abuse, to name a few. Some factors may reduce the risk of ROP. Good prenatal care and the use of surfactant, steroids (dexamethasone), and improved neonatal nutritional support may be factors that reduce the incidence and severity of prematurity and ROP. Factors involved in the progression and outcome of ROP are age at threshold (the younger the baby, the more complicated the treatment and the poorer the prognosis); rate of progression (rapid transformation to threshold ROP portends a poorer prognosis); and systemic complications, such as sepsis. Almost all stages 1 and 2 undergo complete resolution. The risk of blindness in infants with stage 3 ROP is reduced with laser37 or lens-sparing vitrectomy,41 respectively (see Chapter 20). The goal of management is to prevent stage 5 ROP; however, retinal reattachment in some cases of stage 5 ROP may offer improved visual function. Mild ROP does not appear to have an impact on visual acuity; however, severe ROP may be associated with impaired visual development and greater risk of high myopia,42 which emphasizes the necessity of periodic monitoring of early visual acuity in infants with a history of ROP. Even after ROP has regressed, later visual changes, such as myopia, anisometropia, amblyopia, and late retinal detachment,43 may develop. Follow-up with a pediatric ophthalmologist and a retina specialist is recommended for many patients with residual regressed ROP. Generally, the premature infant demonstrates few or no symptoms. Therefore, screening is crucial. Later symptoms may indicate that damage has already occurred. Exotropia may indicate an ectopic fovea; leukocoria may indicate a retinal detachment. The AAO and the American Academy of Pediatrics (AAP) recommend that infants with a birth weight less than 1500 g or a GA of 28 weeks or less, as well as selected infants between 1500 and 2000 g with unstable clinical courses, undergo screening examinations.39 The initial examination is often at 4 to 6 weeks of age, regardless of GA.39 The findings should be recorded according to the ICROP study, indicating stage, zone, and clock hour extent of disease, as well as whether Plus disease is present.33 Because of the high medicolegal risk to those who examine for complications of ROP, a protocol for examination should be used, and follow-up of infants is needed; this follow-up should involve the hospital, the neonatal unit, the neonatologist, and the ophthalmologist. The family should be given instructions about the importance of repeat examinations and information about the risks of ROP. The ideal screening should start in the Neonatal Intensive Care Unit (NICU) and follow NICU protocol. Staff and parents have to be informed about the diagnosis and the infants followed up closely. The ophthalmologist should meet with the staff and neonatologist at each NICU to set up a protocol for dilating drops and sterilization of lid specula and scleral depressors. Screening is described in Chapter 20 (also see Fig. 20–11). During the examination, the vital signs of the infant must be monitored. All findings should be described in the chart using ROP forms. The parents are informed after every examination about follow-up and all possible treatments. Follow-up appointments must be scheduled prior to transferring or discharging the infant. Parents must also be informed about the importance of follow-up examinations on longterm visual acuity. Current therapy for ROP, other than preventing prematurity and judicious neonatal management, consists of cryotherapy and laser therapy. The CRYO-ROP study revealed that visual acuity was better in the threshold eyes treated with cryotherapy than in the control eyes:44.4% versus 62.1%.36 Cryotherapy-treated eyes had a lower risk of an adverse anatomic outcome (27.2%) than did control eyes (47.9%).36 The STOP-ROP study, as well as other studies, demonstrated success after laser treatment.37 Argon and diode indirect ophthalmoscopic laser systems are becoming more popular because of portability and ease of use. In addition, laser is associated more with iris inflammation, breakdown of the blood-retinal barrier, and later myopia than is cryotherapy. The entire avascular zone is treated with tightly placed laser spots, within one half to one spot size apart or with contiguous cryotherapy. Eyes are monitored closely after treatment (see Chapter 20). Despite timely treatment, vision can be reduced secondary to retinal detachment, posterior pole hemorrhage, high myopia, and strabismus. Ongoing trials include the Early Treatment for Retinopathy of Prematurity Study, this evaluates whether treatment of some prethreshold disease will result in better visual and anatomic outcome than current management. In the Photo-ROP Study, images obtained from a wide-angle contact camera can be used to screen and monitor.44 Hydroxychloroquine (Plaquenil) is used to treat connective tissue diseases, including systemic lupus erythematosus (discoid, subacute, cutaneous and systemic), rheumatoid arthritis, Sjögren syndrome, polymorphic light eruption, and porphyria cutanea tarda, among others.45,46 It is effective for long-term maintenance therapy, has a modest incidence of toxicity over time, and can be easily combined with multiple chemically unrelated drugs.47–49 Generally well tolerated, hydroxychloroquine may cause retinopathy, keratopathy, and myopathy. Exposure to light amplifies the risk of retinopathy in patients treated with anti-malarials. (Dark glasses are recommended for patients who spend much time in sunlight.) The concentration of hydroxychloroquine in plasma and tissue is directly related to daily dosing. The highest concentration is found in melanin-containing tissues, particularly the choroid, ciliary body, and retinal pigment epithelium (RPE) of the eye.50,51 Therefore, degeneration of the RPE and its effects on the neurosensory retina are major concerns. Numerous reports indicate a low incidence of retinal toxicity with hydroxychloroquine therapy. The chance of occurrence still exists and appears to depend on dosage.52 MacKenzie suggests a daily dosage threshold for retinopathy below which the drugs are safe. He has shown that overdosage might occur with a standard dose (400 mg per day) if lean body weight is not taken into account. MacKenzie detected no retinopathy in more than 900 patients treated with less than 6.5 mg per kilogram of lean body weight of hydroxychloroquine, even when total doses exceeded 1000 g.53 Rynes and colleagues reported the results of a prospective, 7-year follow-up study of 99 patients treated with 400 mg daily.54 Only four patients developed evidence of early toxicity, which was reversible when therapy was discontinued. They found no relationship between toxicity and total dose. In a study of 1500 patients, Morsman and colleagues found only five cases of toxicity; only one of these patients suffered visual loss.45 Levy found one case of retinopathy of 1207 patients, and this occurred in a patient treated with high doses (6.98 mg/kg daily) for more than 7 years.55 The only cases of definite toxicity at doses less than 6.5 mg per kilogram daily of hydroxychloroquine are reported by Mavrikakis and colleagues, who reported two well-documented cases of hydroxychloroquine retinopathy in patients treated for 6.5 and 8 years without exceeding 6.5 mg per kilogram of body weight daily.56 The risk of retinal toxicity for long-term use of hydroxychloroquine has been estimated to be between 0 and 4%.57–59 These studies varied considerably with respect to case definition, sample size, and duration of follow-up. The largest study reported an incidence of retinal toxicity as 0.08% over a median period of 3.3 years.55 Overall, hydroxychloroquine toxicity usually does not occur with daily doses below 6.5 mg per kilogram of body weight of lean body weight and continued longer than 10 years.57 It is generally recommended that regular ophthalmic examinations (every 6 months) be done to detect its occurrence and prevent progression. The earliest sign of toxicity, which may occur before the development of any fundus abnormality, is paracentral visual field loss. The earliest alterations occur in the parafoveal area.59–61 The patients describe their visual changes as distortion rather than blurriness, and the use of the Amsler grid may be helpful in detecting these early field defects.59 If the process progresses, the RPE alterations extend into the foveolar area, and the patient will lose central vision. This would be most noticeable while reading or driving, but patients simply may complain of difficulty in focusing. It is helpful to know reversible side effects of hydroxychloroquine toxicity. These include corneal deposits (verticillata), loss of foveal reflex, and impairment of accommodation.46,51 Corneal deposits are the most frequent observed lesions. They are dose related, often asymptomatic, and not generally considered a contraindication to continuing treatment.47,58,62 Loss of the foveal reflex is considered a sign of early reversible maculopathy or premaculopathy.63 Reevaluation and temporary reduction in dose should be considered. Retinopathy is the major and most potentially serious irreversible side effect of hydroxychloroquine treatment. Initially, mild pigment stippling of the macula is seen on ophthalmoscopy, and, with time, may result in the classic appearance of bull’s-eye maculopathy. On fluorescein angiography, it will appear as a central zone of irregular pigmentation surrounded by a concentric horizontally oval zone of hypopigmentation.47,50,59,61 If the process progresses, peripheral retinal alterations will be noticeable. Easterbrook and Bernstein suggest that maculopathy should be defined as the presence of reproducible bilateral visual field defects.62 These visual-field defects should be present on both Amsler grid and automated visual-field periphery testing. If the patient does have transient or unilateral field defects, he or she does not likely have hydroxychloroquine maculopathy and should not have the therapy discontinued. Overdose can increase the risk of retinopathy. Overdose may be avoided by using an ideal (lean) body weight instead of using the actual weight of the patient when determining daily dosage.47 If an actual body weight is used for obese patients, patients will be overdosed. Hydroxychloroquine distributes differently through body tissues and is not absorbed well by fat, brain, or bone.47,49,53 Tissue distribution of drug from areas of lowest absorption to areas of highest absorption is bone, fat, brain, muscle, eyes, heart, kidneys, liver, lungs, spleen, and adrenal glands. Patients with poor renal function may be overdosed. About 45% of the drug is renally excreted in unchanged form, 8 to 12% is excreted in the feces, and about 5% is sloughed off through the skin.53 Up to 50% of hydroxychloroquine is biotransformed and broken down, and 30 to 60% of it occurs in the liver. Poor liver function is an additional factor that can contribute to overdose.47,50,53 An initial examination is necessary on initiation of hydroxychloroquine treatment. However, retinotoxicity is not diagnosed with less than 9 months of administration, regardless of the dose.62 If treatment is continued more than 6 months, an ophthalmic examination should be performed regularly. The patient should be questioned about visual changes because he or she may notice problems when driving or reading. It is recommended that the Amsler grid be used as a monthly self-test for patients.59,62 It should be confirmed that the patient has had no change in weight, and renal and hepatic function should be established.49,51 A baseline examination should include distance and near visual acuity testing in each eye; slit-lamp examination of the cornea and retina; visual-field testing, including automated perimetry (small paracentral scotomas within 10 degrees of fixation appear to be a consistent finding in early maculopathy and correspond to pigmentary changes in the fovea.); and baseline photographs of the macula in both eyes. Patients should be monitored every 6 months; if no symptoms or signs are revealed, it is safe to continue treatment. Any patient taking hydroxychloroquine in any amount and for any length of time who complains of a disturbance in central vision and has signs of retinal toxicity (such as paracentral scotoma, pigmentary changes, or bull’s-eye maculopathy) must discontinue therapy and have follow-up for progression (Fig. 21–2). Severe retinopathy may cause progressive visual loss even after discontinuation of therapy.49,50,59,61 Hydroxychloroquine toxicity is rare but serious and often progressive; thus, physicians caring for such patients must be vigilant. The Age-Related Eye Disease Study (AREDS) is a randomized, placebo-controlled, double-masked clinical trial to study the effects of antioxidants or zinc, in combination or alone, on the risk of age-related macular degeneration (AMD) or age-related cataract.66–70
Screening for Specific Diseases
Diabetic Retinopathy
What Is Diabetic Retinopathy and Why Screen for It?
Who Is at Risk for Diabetic Retinopathy?
What Is the Pathophysiology of Diabetic Retinopathy?
What Are the Types of Diabetic Retinopathy, and How Are They Managed and Treated?
NONPROLIFERATIVE DIABETIC RETINOPATHY
ESTIMATING THE RISK OF PROGRESSION OF DIABETIC RETINOPATHY
PROLIFERATIVE DIABETIC RETINOPATHY
What Are the Symptoms of Diabetic Retinopathy?
When Should a Diabetic Patient Have Follow-up Eye Examinations?
When Should Pregnant Women Be Screened for Diabetic Retinopathy?
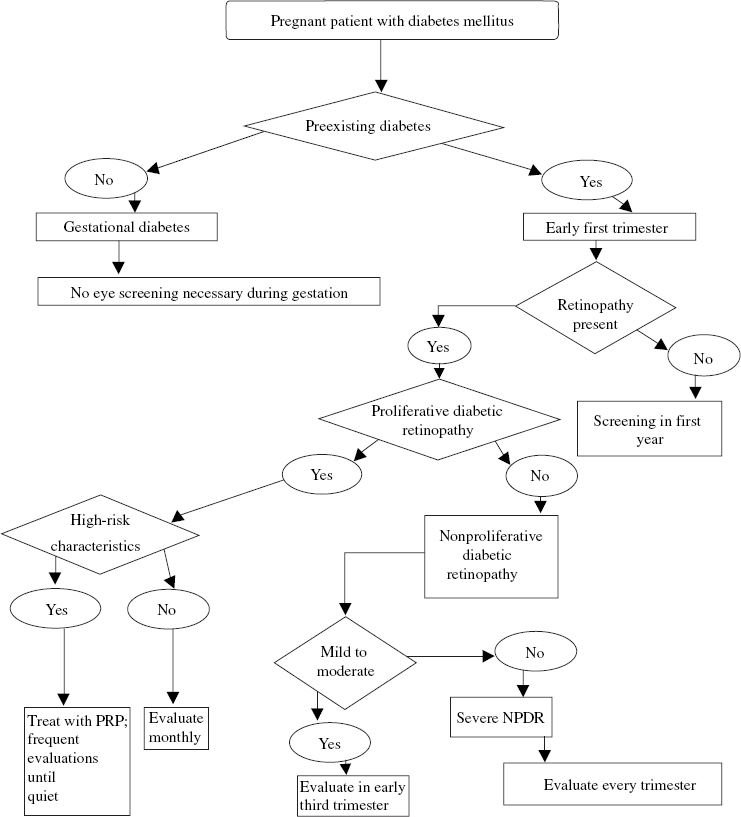
Who Is at Risk for Blindness from Diabetic Retinopathy?
Human Immunodeficiency Virus (HIV) and Retinal Disease
For What Retinal Diseases Should the HIV Patient Be Screened?
What Is HIV Retinopathy?
Cytomegalovirus Retinitis
WHO SHOULD BE SCREENED FOR CMV RETINITIS?
HOW DOES CYTOMEGALOVIRUS RETINITIS PRESENT?
WHAT ARE THE SYMPTOMS OF CMV RETINITIS?
When Should an HIV Patient See an Ophthalmologist?
HOW IS CYTOMEGALOVIRUS RETINITIS TREATED?
WHAT IS IMMUNE RECOVERY VITREITIS?
Retinopathy of Prematurity
What Is Retinopathy of Prematurity and Why Screen for It?
How Is the Severity Classified?
Who Is at Risk?
Who Is at Risk for Blindness from Retinopathy of Prematurity?
What Are the Symptoms of Retinopathy of Prematurity?
When Should a Retinopathy of Prematurity Patient Undergo Screening?
How Is Screening Performed?
How Is Retinopathy of Prematurity Treated?
Hydroxychloroquine Toxicity
What Is Hydroxychloroquine?
Why Screen for Hydroxychloroquine Toxicity?
What Are the Signs and Symptoms of Hydroxychloroquine Toxicity?
How Can Hydroxychloroquine Toxicity Be Prevented?
How Is Hydroxychloroquine Toxicity Examined and Treated?
The Age-Related Eye Disease Study
Ento Key
Fastest Otolaryngology & Ophthalmology Insight Engine