Class I—No CFTR protein produced
Class II—CFTR protein is degraded in the cell before reaching the plasma membrane
Class III—CFTR protein reaches the plasma membrane; however, is unable to be activated
Class IV—CFTR protein reaches the plasma membrane and is activated; however, it does not properly conduct ion flow
Class V—CFTR protein produced in decreased amounts
Table 9.2
Brief summary evidence of affected organ systems with CF (JPeds 2008)
Upper airway |
– Chronic rhinosinusitis |
– Nasal polyps (highly suggestive of CF-sweat test should be performed) |
Lower airway |
– Chronic cough |
– Recurrent pneumonia/infection with “CF specific” bacteria-P. Aeruginosa, Methicillin Resistant S. Aureus, non-typeable H. Influenzae; S. maltophilia, B. cepacia |
– Radiographic abnormalities (bronchiectasis, mucus plugging) |
– Evidence of airway obstruction on pulmonary function testing or clinical examination |
– Digital clubbing |
Pancreas/GI tract |
– Failure to thrive, steatorrhea (due to malabsorption secondary to pancreatic insufficiency) |
– Elevated liver function tests, abnormal bile transport |
– Deficiency of fat soluble vitamins (Vitamin A, D, E and K) |
Reproductive tract |
– Absence of vas deferens |
Epidemiology of Sinus Disease in CF
Several studies have highlighted the lack of nasal sinus symptoms in individuals with CF. Therefore, estimates of the incidence of nasal sinus disease vary, with figures as high as 71–100 % [5]. To address concerns about the accuracy of estimates of sinus disease in CF patients, many experts advocate using questionnaires such as the Sinonasal Outcome Test-22 (SNOT-22) to improve both the ability to diagnose sinus disease and more objectively quantify the response to therapies/surgical intervention [6]. However, these scoring systems have not been well validated in patients with CF [7]. A recent study in the Journal of Cystic Fibrosis [8] demonstrated that sinus disease is more severe in patients with class I–III mutations. This was based on CT scan findings, endoscopy and a symptom scoring system providing support for a previously hypothesized relationship between CFTR genotype and sinus disease phenotype.
Relevant Anatomy and Pathophysiology
The paranasal sinuses are a series of paired mucosa-lined bony chambers that surround and drain into the nose. They are named for the bones into which they have pneumatized: maxillary, ethmoid, frontal and sphenoid. The maxillary and ethmoid sinuses are present though diminutive at birth. The maxillary sinuses expand from their initial outpouching of the middle meatus at birth and continue to grow into early adulthood. The anterior ethmoids emerge from the middle meatus as well and expand into the ethmoid bone throughout childhood and into early adulthood. The frontal sinuses eventually extend as an outgrowth of the anterior ethmoids into the frontal bone. They are not usually radiographically evident until age 6–7 years; their growth continues into adulthood. The sphenoid sinuses are usually not visible on radiographs until about age 1 year. They form through marrow degeneration within the sphenoid body and they fully develop fully by late adolescence [9]. Each sinus generates mucus, which it drains via a characteristic drainage pathway into the nose. This mucociliary clearance occurs through the action of ciliated epithelial cells that line the nose and sinuses. An understanding of these pathways is important, as their obstruction is felt to be a key factor in the pathogenesis of chronic rhinosinusitis (CRS).
The frontal, maxillary and anterior ethmoid sinuses share a common pathway of drainage into the middle meatus through an anatomical region known as the ostiomeatal complex . The posterior ethmoid and sphenoid sinuses drain into the superior meatus via the sphenoethmoidal recess. Ultimately, the products of the sinonasal mucus-producing cells are swept posteriorly into the pharynx and swallowed. A healthy adult nose generates up to 1 L of mucus per day. In the healthy state, the amount, viscosity and composition of mucus allow it to be effortlessly circulated through the nose.
The increased viscosity of mucus produced in patients with CF can lead to mechanical obstruction of sinus ostia and mucus stasis. Consequently, local infection and tissue hypoxia then develop and generate additional inflammation which perpetuates a vicious cycle that compounds the impairment of mucociliary clearance and is a fertile environment for subsequent bacterial colonization within these regions of diminished blood supply [10]. Another consequence of obstructed mucus flow is mucocele formation, whereupon an expanding collection of mucus exerts pressure on the bony confines by which it is trapped. Mucoceles have the ability to erode into neighboring structures such as the orbit or cranium (Fig. 9.1).
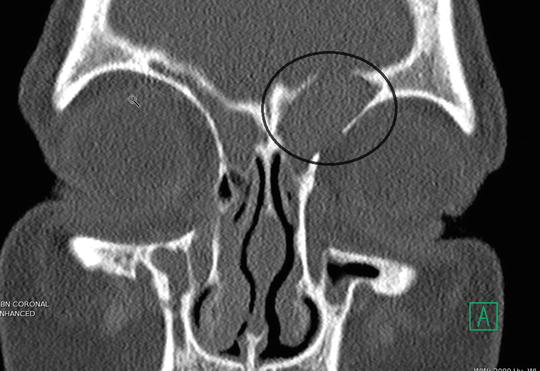
Fig. 9.1
The black oval highlights a left expansile frontoethmoidal mucocele which is eroding into the orbit and intracranially
Nasal polyps (NP) are present in up to 50 % of CF patients and their prevalence is highest during adolescence [11–13]. Since polyps are otherwise rare in children, their presence should alert clinicians to the possibility of CF being present. Conversely, many CF patients do not have polyps, underscoring the need for vigilance on the part of clinicians to suspect CF when appropriate (Table 9.2) even if polyps are absent. The pathogenesis of sinonasal polyposis is unknown although the presence of chronic inflammation clearly plays a role. The inflammatory cellular milieu associated with NP appears to be different in patients with CF when compared with non-CF patients who have polyposis associated with chronic rhinosinusitis (CRSwNP). In patients with CF, polyps are more often associated with a neutrophilic infiltrate whereas eosinophilic associated inflammation is more commonly seen in patients with CRSwNP [11]. Nevertheless, NP are grossly identical in CF and non-CF patients as are their effects of nasal obstruction and impedance of normal mucus flow.
Radiographically, the classic sinonasal anatomic aberrance associated with CF is hypoplastic sinuses with medialization of the lateral nasal walls from expansile polyps within the maxillary antra [14–21] (Fig. 9.2). The maxillary and ethmoid sinuses are commonly smaller than in non-CF patients and the frontal and sphenoid sinuses are sometimes absent (Fig. 9.3). The mechanism for this developmental hypoplasia is not fully elucidated and there is controversy as to whether it develops as a consequence of a patient’s genetic abnormality or from chronic infection itself. It has been stated that normal developmental pneumatization is impaired as a result of chronic inflammation and reduced ventilation alone as is seen in the temporal bone in patients who suffer early chronic otitis media [13]. However, Kim et al. [9] noted that patients with chronic rhinosinusitis without CF did not suffer from the same degree of sinonasal hypoplasia that is seen in CF patients. In a study examining the correlation of genotype with the degree of sinus hypoplasia, Woodworth et al. [22] found that homozygosity for the delta F508 mutation of the CFTR, the most common genotype, was predictive of more severe hypoplasia. A mechanism by which such a mutation would generate this phenotype remains unidentified. Externally, the skeletal changes that can occur due to expansile polyp formation can lead to externally visible stigmata, such as hypertelorism or proptosis [23].
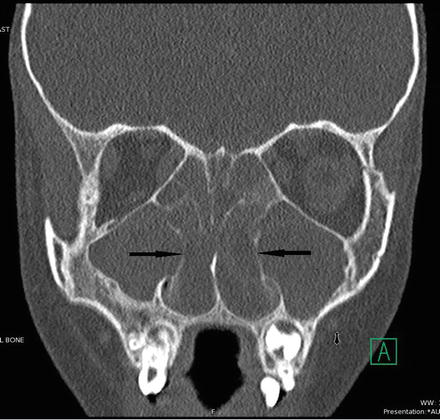
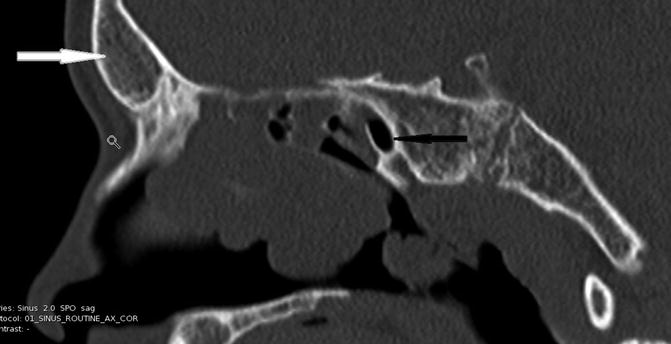
Fig. 9.2
Bilaterally maxillary polyps with overall small maxillary sinuses and medialized infundibula (black arrows)
Fig. 9.3
Absent frontal (white arrow) and hypoplastic sphenoid (black arrow) in this 15 year-old patient with CF
Clinical Presentation
As mentioned previously, sinus disease symptoms are not always elicited on routine history taking. However, reported symptoms include nasal congestion (81 %), rhinorrhea (>50 %), headache (51 %), difficulties with smell/taste (27–66 % depending on the study), morning cough and constant throat clearing [24].
Physical Examination
Chronic rhinosinusitis in CF has been defined as inflammation of nose/paranasal sinuses for >12 weeks with (two or more symptoms): nasal blockage, obstruction, congestion, nasal discharge, facial pain/pressure, and/or reduction in olfaction [25]. One must additionally have at least one of the following finding on examination-nasal polyps, mucopurulent discharge, edema/mucosal obstruction or mucosal changes. Nasal examination begins with simple observation of the patient and should be accompanied by a complete head and neck exam. Patients should be observed for evidence of nasal obstruction such as open mouth or stertorous breathing. Hypertelorism or subtle proptosis suggests underlying expansile mucocele or polyps. Intranasal exam begins with anterior rhinoscopy, which can be accomplished using an otoscope fitted with a wide (>4 mm) speculum or by using a nasal speculum. Polyps, if present, are usually evident through this method (Fig. 9.4). Attention should be directed to the middle meatus from which polyps typically arise. Mucopurulent discharge seen in this region suggests active sinonasal infection. Nasal endoscopy can provide further information about the presence of polyps, purulence and the degree of apparent inflammation present within the nasal cavity. Note that while a polyp is a protuberant growth of solid tissue that arises from nasal mucosa and is visible; a mucocele is a sequestered collection of mucus within a sinus, without a drainage route and therefore is not visible on exam. Their presence is detected either coincidentally on imaging or because infection of their contents causes acute symptoms or external signs (like proptosis). Nasal endoscopy may not be well tolerated in young children but can be accomplished with minimal discomfort. Use of child life specialist or distraction techniques will significantly aid in cooperation with this examination.
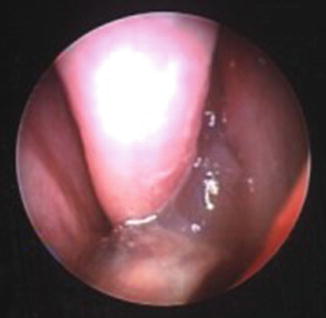
Fig. 9.4
Nasal polyps emanating from the left middle meatus
Radiographic Studies
CT imaging is considered the gold standard imaging modality for evaluating the paranasal sinuses due to its unparalleled resolution of air, bone and soft tissue. However, since CF patients essentially always have abnormalities on sinonasal CT and since these abnormalities do not predictably correlate with symptoms, CT is of little value when performed simply to establish the presence or absence of sinusitis. Since CT, particularly when performed in children, may increase future risk of leukemia and brain cancer [26], clinicians must consider their likelihood for improved management prior to obtaining them. CT scans should be performed for preoperative evaluation prior to endoscopic sinus surgery, after the decision to proceed with surgery has been reached clinically. Non-contrast images with reconstructed coronal views should be ordered. Requesting contiguous 1 mm axial images is prudent since this degree of resolution and method of scan acquisition is necessary for image guided surgery. Additionally, CT imaging is indicated in patients in whom a complication of acute exacerbated sinusitis, such as orbital or intracranial extension of infection, is suspected. In these cases, the addition of IV contrast is necessary to assess for such a complication.
Microbiology of the CF Sinus
Chronic rhinosinusitis of infectious etiology in the CF patient is due predominately to bacterial pathogens with few reports of disease due to fungi such as Aspergillus species. Recent data support the hypothesis that colonization and infection of the sinuses by bacteria is a reservoir for colonization of the lower airway and pulmonary infection [27]. It is also possible that bacteria from the lower airway can move anterograde and infect the sinuses as well. Evidence for this close relationship between bacteria chronically colonizing the sinuses and lower airway infection results from studies that compare bacteria cultured from the sinuses with cultures from the lower airway of the same patient. While the organisms recovered vary depending on the study and the individual patient, a consistent finding is that the traditional CF bacterial pathogens of the lower respiratory tract are commonly found in the sinuses as well [27–31]. For example, two of the most common organisms found in the sinuses of CF patients are Staphylococcus aureus, ranging from 46 % of studied patients [28] to 71 % [27], and Pseudomonas aeruginosa, 27 % in one study [27] and 57 % in another [28] (including mucoid strains). The recovery of Haemophilus influenzae, Streptococcus pneumoniae and other pathogens of the lower respiratory tract is also reported [27–31].
Bacteria not necessarily implicated in sinus disease such as viridans group streptococci and Neisseria sp. can still be present in sinuses from both CF and healthy patients. A more detailed genotypic/phenotypic examination of P. aeruginosa strains recovered from the sinuses and lungs of the same patient with CF demonstrate similar adaptive mutations and phenotypes in both the sinuses and the lung despite differences in the microenvironment [10, 31]. While P. aeruginosa form biofilms in the lungs and sinuses, lung infection is associated with a robust neutrophil response, while in the sinuses a strong IgA response blunts neutrophil recruitment. Despite these differences, P. aeruginosa recovered from both the lungs and sinuses exhibited decreased motility and protease activity compared with environmental strains [31]. After lung transplant, lower respiratory infection with strains present prior to transplant is reported, again arguing that bacteria from the upper airway are a source of lower airway disease.
More recently, deep DNA sequencing molecular techniques have been applied to characterize the bacterial populations that live in different ecological niches of the human body (the human microbiome). Results from multiple studies demonstrate that bacterial culture is an insensitive technique to adequately capture the microbial diversity that colonizes the human body, including the bacterial population in the sinuses. Many organisms, especially anaerobes, do not grow well under standard conditions in the clinical microbiology laboratory leading to an underestimate of the numbers and types of these organisms present. While comprehensive data are lacking, molecular evidence is emerging that sinuses of CF patients are populated with anaerobes such Propionibacterium acnes [27]. Since these organisms can also be found in sinus samples from non-CF patients, the contribution of these uncultivated organisms to pathogenesis and sinus disease in CF patients remains unclear. It does raise the interesting possibility that unrecognized pathogens exist in the CF sinus that contribute to disease but currently little data exists to support this hypothesis.
Initial Therapeutic Options
Nasal irrigations are commonly used to treat nasal sinus symptoms in patients with CF to remove thick secretions and improve mucociliary clearance of the nasal passages. Saline irrigation solutions may be composed of isotonic or hypertonic with/without bicarbonate solutions to provide optimum “clearance”. Devices vary from squeeze bottle to neti pot to even home electronic irrigation devices. There are no large randomized controlled trials in CF patients to support this approach. However, it is a widely accepted method of maintaining good sinus health both pre/post-surgical intervention.
Topical nasal steroids have also been used in CF to decrease nasal inflammation and decrease size of nasal polyps. There is a single randomized controlled trial of betamethasone which demonstrated both decrease in polyp size as well as nasal symptoms during treatment [32]. In clinical practice, some have combined both topical nasal steroids (i.e. budesonide) mixed into saline irrigation to maximize lasting topical benefit but large supporting clinical trial data have not been obtained.
Another therapy currently under investigation is dornase alfa which enzymatically degrades extracellular DNA (a large component of the debris in CF mucus). Inhaled into the lower airway, Dornase alfa is an effective medication for maintaining lower airway function and decreasing the frequency of pulmonary exacerbations in individuals with CF. It is a recommended daily therapy in both the US and Europe for patients over the age of 6 [33]. Several studies have suggested dornase alfa can improve upper airway function and sinus disease based on symptoms as well as endoscopic evaluation. Furthermore, a recent study by Mainz et al, demonstrated significant improvements in pulmonary function as well [34]. This method of aerosoling dornase alfa by intranasal nebulization to deliver medication the sinuses is not widely available in the US currently.
The latest in therapy for patients with CF are a variety of medications that work as correcting the underlying molecular defect. These drugs, known as correctors and potentiators, are mutation specific. The most widely known drug, ivacaftor, corrects the defect associated with class III mutations. There are several case reports of patients who have significant improvement in their upper airway symptoms and sinus CT once starting the medication [35, 36].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


