Purpose
To evaluate the evidence supporting a role for senescent changes in cerebrospinal fluid (CSF) circulatory physiology in the pathogenesis of normal-tension glaucoma (NTG).
Design
Literature review and personal perspective of the authors.
Methods
Analysis of selected articles in the peer-reviewed literature with interpretation and perspective.
Results
Recent studies have reported that intracranial pressure is lower in patients with NTG when compared with patients with primary open-angle glaucoma and nonglaucomatous control subjects. It has been suggested that a low intracranial pressure in patients with normal intraocular pressure could lead to glaucomatous damage. This low intracranial pressure, leading to an abnormally high trans–lamina cribrosa pressure difference, could result in barotraumatically induced optic nerve damage at the lamina cribrosa. However, several experimental studies do not support the speculation that low intracranial pressure and the resulting pressure-dependent effects cause bowing back of the lamina cribrosa and optic disc cupping. On the other hand, CSF production and turnover have been shown to be decreased in aging and in pathologic conditions, such as Alzheimer disease and normal pressure hydrocephalus. Interestingly, recent studies have revealed that both Alzheimer disease patients and patients with normal pressure hydrocephalus may have a higher risk of developing glaucoma. Therefore, we believe that CSF circulatory failure, ultimately resulting in reduced neurotoxin clearance along the optic nerves, could be an alternative explanation as to why glaucoma develops in patients with low intracranial pressure.
Conclusions
On the basis of the evidence available from the peer-reviewed literature, our tentative conclusion is that age-related changes in CSF circulatory physiology and the subsequent decrease in CSF turnover, with diminished clearance of toxic substances, can account for, at least in part, the pathogenesis of NTG. It should be stressed that for the moment at least, the present hypothesis remains unproven. Further research will be necessary to determine the possible role of CSF circulatory dysfunction in NTG. If confirmed, this hypothesis could provide new, important insights into the pathogenesis of NTG.
Glaucoma is one of the leading causes of irreversible blindness. Primary open-angle glaucoma (POAG), the most common type, is a progressive optic neuropathy characterized by optic disc cupping and functional changes in the visual field. It becomes more severe with advance of age. In the glaucomatous optic nerve, cupping of the optic disc reflects a loss of retinal ganglion cell axons and a posterior bowing of the lamina cribrosa, accompanied by extensive remodeling of the optic nerve head. Although elevated intraocular pressure (IOP) remains one of the most important risk factors for POAG, it was reported that approximately 20%-90% of patients with POAG, with percentages seeming to vary according to race, have normal IOP measurements, a condition known as normal-tension glaucoma (NTG). The pathophysiology of NTG remains unclear. In contrast to high-tension glaucoma, in which IOP plays a more significant role, non-IOP factors such as hemodynamic abnormalities or vasospastic events might be more critical in NTG. In addition to vascular risk factors, it has been suggested that a low intracranial pressure in patients with normal IOP could lead to glaucomatous damage. Recent studies reported that intracranial pressure is lower in patients with NTG when compared with patients with POAG and nonglaucomatous control subjects. The mechanisms most commonly proposed to explain the contribution of low intracranial pressure and thus high trans–lamina cribrosa pressure difference (the difference between IOP and pressure in the subarachnoid space of the optic nerve) to glaucoma are direct strain on the lamina cribrosa and impairment of axonal transport. However, several experimental studies do not support the speculation that low intracranial pressure causes bowing back of the lamina cribrosa and optic disc cupping. Moreover, the role of axoplasmic flow stasis in the development of glaucomatous optic neuropathy has been questioned.
In the present paper, we propose an alternative hypothesis according to which the lower cerebrospinal fluid (CSF) pressure reported in NTG patients could be an indicator of CSF circulatory failure (eg, because of reduced CSF production), thereby leading to glaucomatous damage. We hypothesize that changes in the senescent brain may play a fundamental role in the development of some, if not many, cases of NTG. With aging, changes in CSF circulatory physiology result in decreased CSF turnover and CSF stasis. Given the central role of CSF turnover in the clearance of potentially toxic metabolites from the brain, one might expect that CSF circulatory dysfunction could ultimately result in reduced neurotoxin clearance in the subarachnoid space surrounding the optic nerve and lead to glaucomatous damage. Circumstantial evidence supporting this idea comes from studies in pathologic conditions, such as Alzheimer disease and normal pressure hydrocephalus, in which these changes in CSF circulatory physiology are significantly accentuated. Data for this personal view were identified through searches of PubMed with the search terms “glaucoma,” “normal-tension glaucoma,” “intracranial pressure,” “Alzheimer’s disease,” “normal pressure hydrocephalus,” “beta-trace protein,” and “cerebrospinal fluid pressure, production, turnover and clearance.” In addition, other relevant articles were identified from supplemental searches. Only papers published in English between 1980 and 2012 were included. The final reference list was generated on the basis of originality and relevance to the scope of this review.
Presentation of the Hypothesis
The Cerebrospinal Fluid Pressure is Lower in Patients With Glaucoma
Recent research findings suggest the potential pathogenic role of an abnormally low CSF pressure in the development of POAG and NTG. In a retrospective case-control study, Berdahl and associates reported the intriguing new observation that mean CSF pressure as measured by lumbar puncture was 33% lower in a group of 28 patients with POAG than in a control group of 49 nonglaucomatous patients (9.2 ± 2.9 mm Hg vs 13.0 ± 4.2 mm Hg or 124 ± 39 mm H 2 O vs 177 ± 57 mm H 2 O; P < .00005). Subjects were considered to have POAG if they were diagnosed with POAG by a glaucoma specialist, had characteristic optic nerve changes, and had visual field loss consistent with glaucoma. Linear regression analysis showed that cup-to-disc ratio correlated independently with IOP ( P < .0001), CSF pressure ( P < .0001), and the trans–lamina cribrosa pressure difference ( P < .0001). Multivariate analysis demonstrated that larger cup-to-disc ratio ( P < .0001) was associated with lower CSF pressure. The authors noted that their observation supports the concept that an abnormal high trans–lamina cribrosa pressure difference, whether the result of elevated IOP, reduced CSF pressure, or both, plays an important role in glaucomatous optic nerve damage. The lamina cribrosa forms the bottom of the optic cup in the optic nerve head and acts as a pressure barrier between the intraocular pressure space and the retrobulbar CSF pressure space. Normally, the IOP ranges from 10-21 mm Hg, whereas the CSF pressure ranges from 5-15 mm Hg. From a mechanical perspective, a similar posteriorly directed force is caused by either a lower pressure on the CSF side of the lamina or a higher pressure on the intraocular side.
In another study with similar design, Berdahl and associates compared lumbar CSF pressure measurements in 57 subjects with POAG, 11 subjects with NTG (a subset of POAG), and 27 subjects with ocular hypertension (OHT) with 105 age-matched control subjects without glaucoma (66 in the age-matched control group for comparison with POAG and the NTG subset and 39 in the age-matched control group for comparison with the OHT group). The CSF pressure was significantly lower in POAG compared with age-matched control subjects without glaucoma (9.1 ± 0.77 mm Hg vs 11.8 ± 0.71 mm Hg; P < .0001). The subjects with NTG also had a lower CSF pressure compared with the control subjects (8.7 ± 1.16 mm Hg vs 11.8 ± 0.71 mm Hg; P < .01). Furthermore, the CSF pressure was higher in OHT than in age-matched control subjects (12.6 ± 0.85 mm Hg vs 10.6 ± 0.81 mm Hg; P < .05). The mean translaminar pressure difference was 6.1 ± 5.6 mm Hg in POAG and 5.0 ± 4.4 mm Hg in the NTG subset compared with 1.9 ± 4.4 mm Hg in age-matched controls ( P < .05). In the OHT group, the mean translaminar pressure difference was 8.4 ± 5.5 mm Hg compared with 4.4 ± 3.5 mm Hg in age-matched controls ( P < .05). The authors concluded that a reduced CSF pressure may play an important role in the development of POAG and NTG. Conversely, the elevated CSF pressure in OHT may counterbalance the elevated IOP, potentially preventing or slowing glaucomatous optic nerve damage in this patient population.
The findings of Berdahl and associates were recently confirmed by a prospective study that compared lumbar CSF pressure in open-angle glaucoma patients and nonglaucomatous control subjects. The study included 43 patients with open-angle glaucoma (OAG), differentiated into 14 patients with normal-pressure glaucoma and 29 patients with high-pressure glaucoma, and 71 control subjects. The CSF pressure was significantly lower ( P = .013) in the normal-pressure glaucoma group (9.5 ± 2.2 mm Hg) than in the high-pressure glaucoma group (11.7 ± 2.7 mm Hg), in which it was significantly ( P < .001) lower than in the control group (12.9 ± 1.9 mm Hg). The trans–lamina cribrosa pressure difference was significantly ( P < .001) higher in the high-pressure glaucoma group (12.5 ± 4.1 mm Hg) than in the normal-pressure glaucoma group (6.6 ± 3.6 mm Hg), in which it was significantly ( P < .001) higher than in the control group (1.4 ± 1.7 mm Hg). The extent of glaucomatous visual field loss was positively correlated with the height of IOP, negatively correlated with the height of CSF pressure, and positively correlated with the height of the trans–lamina cribrosa pressure difference. The authors concluded that a low CSF pressure in patients with normal IOP could lead to glaucomatous damage.
Age-related Changes to Cerebrospinal Fluid Turnover
The turnover of CSF decreases substantially with aging. This is related to the increase in CSF volume in the brain, the decrease of CSF secretion by choroid plexuses, and the increase in resistance for CSF drainage into the vascular system.
It is believed that CSF is actively produced mainly within the brain ventricles by choroid plexuses. The choroid plexuses are highly vascularized villous structures covered by a single layer of ciliated, cuboid epithelium. Choroid plexuses secrete CSF, synthesize numerous molecules, carry nutrients from the blood to CSF, reabsorb brain metabolism by-products, and participate in brain immunosurveillance. A review by Brown and associates highlighted the molecular mechanisms of CSF production. The epithelial cells of the choroid plexus secrete CSF by a process that involves the transport of Na + , Cl − , and HCO 3 − from the blood to the ventricles of the brain. This creates an osmotic gradient that is accompanied by the secretion of H 2 O. The movement of ions across the cellular membrane is mediated by specific transporters and ion channels that are distributed unequally on the basolateral and apical sides of the choroid plexus epithelial layer. Na + -K + -ATPase, K + channels and Na + -K + -2Cl − cotransporters are expressed in the apical membrane. By contrast, the basolateral membrane contains Cl − -HCO 3 − exchangers, a variety of Na + -coupled HCO 3 − transporters, and K + -Cl − cotransporters. Aquaporin 1 mediates water transport at the apical membrane, but the route across the basolateral membrane is unknown. With aging, choroid plexuses are the site of morphologic changes. There is flattening and loss of the secretory epithelium, basement membrane thickening, lipofuscin accumulation, formation of Biondi bodies, thickening of the fibrous stroma, calcification, and arterial wall thickening. Among the numerous proteins involved in choroidal CSF production, it is known that Na + -K + -ATPase, carbonic anhydrase II, aquaporin 1, and solute carrier family 4, sodium bicarbonate transporter, member 10 are major contributors to CSF secretion. A study by Masseguin and associates showed that aging affects choroidal proteins involved in CSF production. The authors compared choroid plexuses of Sprague-Dawley rats aged 10 or 20 months with those of 3-month-old ones. Progressive and age-related changes in the Na + -K + -ATPase, carbonic anhydrase II, and aquaporin 1 expressions at the apical and/or cytoplasmic level, as suggested by both the decreases in the intensities of immunocytochemical and in situ hybridization signals, indicated that aging decreases notably the protein expression of the enzymes and transporters known to regulate the CSF production in the choroid plexus. The age-related morphologic and enzymatic changes described above are thought to contribute to reduced CSF secretion. Several studies support a trend towards decline as a function of age. In humans, in a newborn, it is 0.60 mL/min, in an adult 0.41 mL/min, and in elderly 0.19 mL/min.
Regardless of evidence for decreased CSF production, there is evidence for reduced CSF turnover with age, caused in part by the increase in CSF volume attributable to moderate brain atrophy. The total CSF volume increases from 140 mL in young adults to more than 300 mL in elderly. Given that CSF turnover is defined as the volume of CSF produced in 24 hours divided by the volume of the CSF space, it decreases significantly with aging. In newborns, young adults, and elderly controls, CSF turnover is 6, 4.5, and 3 times a day, respectively.
A further factor adding to reduced CSF turnover is the age-related increase in resistance to CSF outflow. This is related to fibrosis of meninges and decreased reabsorption.
There is evidence that turnover of CSF helps to clear toxic molecules from the interstitial-fluid space of the brain to the bloodstream. CSF flows from the brain ventricles into interconnecting chambers, namely the cisterns and the subarachnoid spaces, including the subarachnoid space of the optic nerves. Our hypothesis is that senescent changes in CSF circulatory physiology and the subsequent decrease in CSF turnover could ultimately result in reduced neurotoxin clearance in the subarachnoid space surrounding the optic nerve and lead to glaucomatous damage. Some of the evidence that supports this hypothesis comes from studies of diseases in which the CSF turnover is reduced. The age-related changes in CSF circulatory physiology described above are significantly accentuated in pathologic conditions, such as normal pressure hydrocephalus and Alzheimer disease. It has been suggested that both Alzheimer disease and normal pressure hydrocephalus are physiologically related to CSF circulatory failure, resulting in reduced CSF clearance and accumulation of neurotoxins, such as β-amyloid, that play a role in the pathogenesis of Alzheimer disease. Interestingly, recent studies have revealed that both Alzheimer disease patients and patients with normal pressure hydrocephalus may have a higher risk of developing glaucoma.
Cerebrospinal Fluid Circulatory Dynamics in Normal Pressure Hydrocephalus and Alzheimer Disease
In Alzheimer disease, the turnover of CSF decreases and is less than 1.5 times a day. It might even be around 0.75 times a day. This turnover decline is caused in part by the increase in CSF volume in the brain. Brains of patients with Alzheimer disease show atrophy. The turnover time for such an increase in CSF volume is substantially longer. In Alzheimer disease, there is also evidence for a decrease in CSF production. Using the Masserman technique, Silverberg and associates measured a 50% decrease in CSF production among Alzheimer disease patients when compared with Parkinson disease patients. Mean CSF production in Alzheimer disease was 0.20 ± 0.06 mL/min, and in controls 0.42 ± 0.13 mL/min. Structural changes in the choroid plexus coincide with diminished CSF production in Alzheimer disease. Choroid plexuses present similar, although much more pronounced, abnormalities than those observed in aging. In another study, Silverberg and associates reported on cerebrospinal fluid pressure in patients with Alzheimer disease. This study showed a high occurrence of very low CSF pressure among patients with Alzheimer disease. Given that intracranial pressure depends on cerebral tissue volume, cerebrospinal fluid volume, and cerebral blood volume, the marked reduction in CSF production observed in Alzheimer disease patients could be the cause of the lower CSF pressure. Theoretically, the cerebral atrophy in Alzheimer disease might lead to a further reduction of the CSF pressure.
Normal pressure hydrocephalus is also characterized by a lowered CSF turnover. It is lowered to 1.5 times a day, as in Alzheimer disease. It is generally assumed that normal pressure hydrocephalus is a disorder of decreased CSF absorption. During the initial stage of the disease, intracerebroventricular pressure may increase, thereby leading to ventricular enlargement with stretching of the periventricular parenchyma. As the ventricles enlarge, cerebrospinal fluid pressure returns to normal so that it is within the normal range at lumbar puncture. However, the name of this condition is misleading because continuous intracranial pressure monitoring demonstrates intermittently raised intracranial pressure in association with B-waves. B-waves are slow and rhythmic oscillations in intracranial pressure with a period of 0.5-2 minutes. In normal pressure hydrocephalus, there is evidence for CSF stagnation with decreased clearance of various macromolecules. Although the primary change in normal pressure hydrocephalus is an increase in CSF outflow resistance, decreased CSF production also has been reported. In a study using the Masserman technique, CSF production was significantly lower among patients with normal pressure hydrocephalus (0.25 ± 0.08 mL/min) than in age-matched Parkinson disease patients (0.42 ± 0.13 mL/min) or patients with acute hydrocephalus (0.40 ± 0.13 mL/min). Changes in the choroid plexus have also been characterized in normal pressure hydrocephalus. Both elevated CSF outflow resistance and impaired CSF production lead to a decrease in CSF turnover and, in turn, a decreased clearance of macromolecules. In normal pressure hydrocephalus, a decrease in clearance of β-amyloid and tau, the 2 hallmark proteins in Alzheimer disease, is suggested by the higher-than-expected coincidence of Alzheimer disease pathology in cortical biopsy samples obtained at shunt implantation. The coincidence of Alzheimer disease neuropathology among patients with normal pressure hydrocephalus varies from 25%-75% depending on the severity of the clinical dementia.
High Occurrence Rate of Glaucoma Among Alzheimer Disease Patients and Patients With Normal Pressure Hydrocephalus
Studies have identified a higher rate of glaucoma among Alzheimer disease patients and patients with normal pressure hydrocephalus. In a nursing home–based study in Germany, Bayer and associates studied 112 patients with Alzheimer disease and 116 control subjects. The prevalences of glaucoma were reported to be 25.9% in patients with Alzheimer disease and 5.2% in the control group. The diagnosis of probable glaucoma required at least 1 of the following 2 criteria: a characteristic pattern of glaucomatous visual field loss and/or a cup-to-disc ratio of 0.8 or greater with an optic nerve head appearance consistent with glaucoma. In no instance was the diagnosis of glaucoma dependent on the level of IOP. The mean IOP was significantly different between eyes with and without glaucoma in both groups, the control subjects and the patients with Alzheimer disease. NTG was found in 11 of the 29 glaucoma patients with Alzheimer disease.
In a Japanese study, Tamura and associates found a prevalence of open-angle glaucoma of 23.8% among 172 patients with Alzheimer disease, which was significantly higher than the 9.9% among 176 control subjects. Probable OAG was diagnosed by width of the angle of the anterior chamber >grade 2, a vertical cup-to-disc ratio of the optic nerve head >0.7, and/or difference between the vertical cup-to-disc ratio in the eyes >0.2 with characteristic glaucomatous disc change. In almost all Alzheimer disease patients, no reliable data of visual field could be obtained. In no instance was the diagnosis of OAG dependent on the level of IOP. There was no significant difference between IOPs in Alzheimer disease patients with OAG and without OAG, and almost all Alzheimer disease patients with OAG showed normal IOP (<21 mm Hg). It should be noted that approximately 90% of OAG patients in Japan suffer from NTG.
Contrary to these studies, a longitudinal study investigating the rate of subsequent Alzheimer disease in 11 721 patients with POAG (including NTG) did not find a significantly increased risk of Alzheimer disease compared with the controls. Another study of 69 patients with NTG who were tracked in nationwide registers for a mean follow-up period of 12.7 years did not find that NTG was associated with increased risk of developing dementia/Alzheimer disease compared with the risk for the general population. It has been suggested that patients with glaucoma do not have an increased risk of developing Alzheimer disease, but, by contrast, Alzheimer disease is a risk factor for glaucoma.
Recently, Chang and Singh performed a case-control study to investigate the likelihood of glaucoma in patients with normal pressure hydrocephalus. The authors identified the medical records of 72 cases of normal pressure hydrocephalus and 72 age-matched controls with hydrocephalus treated at Stanford University Hospital between 1996 and 2007. This study found that the prevalence of glaucomatous disease in patients with normal pressure hydrocephalus was 18.1%, which was much higher than that of the age-matched non–normal pressure hydrocephalus controls with hydrocephalus (5.6%). Glaucoma status of the cases and controls was ascertained based on an elicited history of glaucomatous disease, prior or current IOP-lowering therapy, or prior laser and/or surgical therapy for glaucoma. Insufficient information was available to determine the category of glaucoma that cases and controls were found to have.
The Hypothesis as a Bridge Between 2 Theories of Glaucoma Pathogenesis
In the present paper, we hypothesize that age-related changes in CSF circulatory physiology and the subsequent decrease in CSF turnover, with diminished clearance of toxic substances, can account for, at least in part, the pathogenesis of NTG. Such CSF circulatory failure might ultimately result in reduced neurotoxin clearance in the subarachnoid space surrounding the optic nerve and lead to glaucomatous damage. Supportive evidence for this hypothesis comes from the above-noted studies showing decreased CSF turnover in normal pressure hydrocephalus and Alzheimer disease, and indicating that both Alzheimer disease patients and patients with normal pressure hydrocephalus may have a higher risk of developing glaucoma. Moreover, in a recent report, Nucci and associates described a glaucoma patient with medically controlled IOP who experienced disease progression concomitantly with the onset of mild cognitive impairment and positivity for CSF markers of Alzheimer disease (decreased β-amyloid and elevated levels of total and phosphorylated tau). The authors suggested the possibility that altered CSF circulatory dynamics in this case reduced neurotoxin clearance along optic nerves in the subarachnoid space and that deposits/aggregates of tau and/or other toxic molecules may have contributed to the glaucoma progression. Interestingly, Killer and associates hypothesized that NTG may be attributable to sequestration of CSF within the terminus of the optic nerve subarachnoid space, creating a stagnant region accumulating substances toxic to the adjacent optic nerve head. A disturbed CSF exchange between the CSF in the intracranial spaces and the CSF in the subarachnoid space surrounding the optic nerves might result in reduced CSF turnover in the subarachnoid space of the optic nerve, leading to accumulation of potentially toxic substances. The concept of an optic nerve compartment syndrome is supported by several recent studies. Anatomically, the subarachnoid space of the optic nerve resembles a cul-de-sac that has the potential for decreased CSF turnover. Killer and associates suggested that the subarachnoid space surrounding the optic nerve may become an isolated CSF compartment under specific pathologic conditions. Possible mechanisms leading to the development of compartmentation of the subarachnoid space of the optic nerve and subsequently to impaired local CSF turnover include inflammatory changes as well as mechanical stress on the arachnoid and its trabeculae, as well as on glial cells in the optic nerve. The subarachnoid space of the optic nerve (including the trabeculae and septae) is lined with meningoepithelial cells. These cells are known to be highly reactive to various stimuli that can cause proliferation. Proliferation of meningoepithelial cells might result in narrowing of the subarachnoid space surrounding the optic nerves and might lead to CSF compartmentation. The accumulation of biologically active molecules in this blocked optic nerve compartment might further produce a toxic effect on the optic nerve and lead to glaucomatous damage. Moreover, a self-perpetuating cycle could ensue whereby these toxic substances might further harm meningoepithelial cells.
With regard to possible neurotoxic mechanisms, Killer and associates postulated that an accumulation of biologically highly active substances such as lipocalin-like prostaglandin D synthase, also called beta-trace protein, following compartmentation could exercise a harmful effect on axons and mitochondria of the optic nerve, and could adversely affect the blood vessel tone of the pial plexus supplying the optic nerve in the subarachnoid space. The highest concentration of mitochondria is located right behind the lamina cribrosa in nonmyelinated axons. The main function of mitochondria is the production of adenosine triphosphate, which is essential for cell survival. Given that the unmyelinated optic nerve has a high relative demand for mitochondrial enzyme activity, the immediate retrobulbar portion of the optic nerve may be particularly vulnerable to toxic effects. As the biochemical effects of beta-trace protein are several, accumulation of this agent in a blocked optic nerve compartment might, at least theoretically, have a significant effect on the optic nerve. Beta-trace is a mainly brain-derived protein. The main sources of biosynthesis in the brain for this protein are the leptomeninges. It is one of the most abundant locally synthesized proteins in the CSF and plays a significant role in the metabolism and health of the central nervous system. Some of the functions of beta-trace protein seem contradictory. On one hand, it is believed to play a role in neuroprotection. On the other hand, several studies have reported apoptosis-inducing properties of beta-trace in neuronal tissues. Given that astrocytes play a critical role in maintaining the integrity of axon function in the central nervous system and specifically in the optic nerve, Xin and associates investigated the biochemical effects of beta-trace protein on the proliferation of astrocytes and on the production of adenosine triphosphate by astrocyte mitochondria in an in vitro model. The authors demonstrated an inhibitory effect of beta-trace on both proliferation of astrocytes and production of astrocyte adenosine triphosphate. Beta-trace protein is also involved in the synthesis of prostaglandins that participate in the regulation of vascular tone. Obviously, beta-trace is only one of many CSF components with biological activity, and other substances could also be harmful. Clearly, the findings of Killer and associates raise questions concerning the possible biological effects of accumulated CSF and its components on the optic nerve following compartmentation. To address these questions further studies will be needed. To date, there is no experimental evidence that an intact optic nerve can be damaged by accumulated toxins. In order to investigate this further, infusion of such toxins could be performed in the subarachnoid space of the optic nerve as part of a laboratory investigation.
Interestingly, our hypothesis may bring together 2 seemingly quite different theories of glaucoma pathogenesis by interpreting them as sequential steps involved in the development of the disease. Berdahl and associates and others focus on the potential pathogenic role of an abnormally low intracranial pressure in the development of POAG and NTG. This low intracranial pressure, leading to an abnormally high trans–lamina cribrosa pressure difference, could result in barotraumatically induced optic nerve damage at the lamina cribrosa. With regard to the hypothesis presented here, the question is whether pressure-dependent effects are indeed directly involved in the development of glaucoma when intracranial pressure is abnormally low, or whether CSF circulatory failure could be an alternative explanation as to why glaucoma develops in patients with low intracranial pressure. Obviously, both mechanisms might also operate at the same time. First of all, the observation of a greater likelihood of developing glaucoma in patients with normal pressure hydrocephalus supports the notion that CSF stagnation may play a contributory role in the pathogenesis of glaucoma. Indeed, in the setting of normal pressure hydrocephalus, CSF stasis is present in the absence of low intracranial pressure. Furthermore, in recent years, a number of objections have been raised against the hypothesis that low CSF pressure could be linked to glaucoma through pressure-dependent effects on the lamina cribrosa. This hypothesis assumes that the trans–lamina cribrosa pressure difference (the difference between IOP and pressure in the subarachnoid space of the optic nerve) is increased in glaucoma patients who have low CSF pressure. This is based on the assumption that the lumbar CSF pressure, which is known to correlate with intracranial pressure, can be extrapolated to the subarachnoid space surrounding the optic nerve. Given that it is not possible to measure the retrolaminar pressure clinically, the above-noted retrospective and prospective studies of CSF pressure in patients with glaucoma took the lumbar CSF pressure measurement as surrogate for pressure in the orbital cerebrospinal fluid space. However, using computed tomography scanning, Jaggi and associates found that patients with NTG have increased optic nerve sheath diameters. Distension of the optic nerve sheath is a typical feature of elevated intracranial pressure, not lower intracranial pressure. Moreover, CT cisternography, which was used in a study of 18 patients with bilateral NTG, demonstrated a significantly higher intracranial density of contrast-loaded cerebrospinal fluid in the intracranial spaces (basal cisterns) compared with the density of contrast-loaded cerebrospinal fluid in the subarachnoid space surrounding the optic nerve. This difference was not present in the small control group without NTG. These findings suggest that the subarachnoid space surrounding the optic nerve can develop into a separate CSF compartment with a different CSF composition, different CSF dynamics, and probably a different pressure in the setting of NTG. Even assuming that the low lumbar CSF pressure in patients with NTG results in an increased trans–lamina cribrosa pressure difference, the question remains whether such fall in CSF pressure could cause bowing back of the lamina cribrosa and cupping. As pointed out by Hayreh, a great deal of literature has shown little or no foundation for the idea that the translaminar imbalance between the IOP and CSF pressure caused by low CSF pressure can cause bowing back of the lamina cribrosa, and consequently glaucomatous optic disc cupping. The lamina cribrosa is a rigid, compact band of connective tissue, anchored firmly to the surrounding sclera and posteriorly to the longitudinal fibrous septa of the optic nerve. Several experimental studies where IOP was acutely raised to 40-60 mm Hg in enucleated human eyes and in primate eyes have shown that even a marked difference between the IOP and CSF pressure caused no ophthalmoscopically detectable change in the lamina cribrosa. In the study by Berdahl and associates, the mean translaminar pressure difference was 6.1 ± 5.6 mm Hg in POAG and 5.0 ± 4.4 mm Hg in the NTG subset compared with 1.9 ± 4.4 mm Hg in age-matched controls—that is, a difference of 4.2 mm Hg in POAG and 3.1 mm Hg in NTG compared with the control group. Berdahl and associates suggested that, even under the influence of a small trans–lamina cribrosa pressure difference, the rigid lamina cribrosa may be capable of slowly remodeling over time, given that it is also a living dynamic structure. Hayreh, however, concluded that the small differences in translaminar pressure between the control and glaucoma patients are not capable of causing any remodeling and bowing back of the rigid, thick compact band of connective tissue of the lamina cribrosa, when even an acutely raised IOP to 40-60 mm Hg produced no ophthalmoscopically detectable change in its position.
However, the fact remains that recent studies reported reduced lumbar CSF pressure and thus lower intracranial pressure in NTG patients when compared with patients with POAG and nonglaucomatous control patients. In any case, this remains an interesting finding and perhaps lends support to our hypothesis of decreased CSF turnover as a contributing mechanism to the development of NTG. Indeed, a decreased intracranial pressure can be the consequence of reduced CSF production. The intracranial pressure is built up by the equilibrium between the production of the CSF by the choroid plexuses in the ventricles of the brain and the outflow of the CSF in the subarachnoid space into the venous blood system. Two genes that are expressed in the choroid plexus and have been shown to be important in CSF production are aquaporin 1 and solute carrier family 4, sodium bicarbonate transporter, member 10. Knockout mouse models of these genes demonstrate a significant reduction in CSF production and intracranial pressure. Therefore, as an alternative to barotraumatic optic nerve damage, we believe that low intracranial pressure could play a role in the pathogenesis of glaucoma through its association with decreased CSF production and turnover. From this point of view, low intracranial pressure could be an indicator of CSF circulatory failure. This CSF circulatory dysfunction could result in reduced clearance of toxic substances in the subarachnoid space surrounding the optic nerve. This, in turn, could lead to compartmentation of the subarachnoid space of the optic nerve. Indeed, it is conceivable that the accumulation of these potentially toxic substances might have an influence on meningoepithelial cells through biochemical mechanisms, eventually leading to increased proliferation and CSF compartmentation. It could thus be postulated that low intracranial pressure could lead to an optic nerve sheath compartment syndrome through its association with CSF circulatory failure. Decreased intracranial pressure, as an indicator of decreased CSF production and turnover, and CSF compartmentation could then be seen as sequential steps in the disease process of NTG.
In line with our hypothesis, CSF proteins increase with age. For example, previous studies reported increased CSF beta-trace concentrations with age. Reduced CSF formation rate and turnover are the most plausible explanation for this phenomenon. CSF proteins like beta-trace protein, which originate primarily from leptomeningeal cells, show a linearly increasing concentration in lumbar CSF in case of pathologically decreasing CSF flow rate. This is the simple consequence of a steady release of the protein into CSF which, because of a slower volume turnover rate, gains a higher protein concentration per volume. Given the age-dependent increase of CSF proteins resulting from CSF circulatory dysfunction, it seems reasonable to assume that some CSF substances may reach concentrations that are toxic to the optic nerve, similar to that which may occur following compartmentation. To assess whether there is evidence in support of our hypothesis, beta-trace concentrations in lumbar CSF could be compared between NTG patients and age- and sex-matched control subjects. A significantly higher lumbar CSF beta-trace concentration in NTG patients compared with controls would support our hypothesis. Certainly, such a study cannot prove causality, it can only provide evidence for an association between NTG and beta-trace protein as a putative causative factor. Moreover, taking the lumbar CSF concentration of beta-trace protein as surrogate for its concentration in the CSF in the subarachnoid space of the optic nerve, it could be argued that lumbar CSF concentrations do not represent the precise concentrations in subarachnoid space CSF. Indeed, apart from the impact of possible compartmentation on subarachnoid space CSF composition, beta-trace protein is produced across the entire leptomeninges and increases in concentration up to 11-fold from the source of CSF production in the ventricles to the lumbar spine. This rostrocaudal increase of beta-trace protein concentration under normal conditions is easily understood as a steady release of beta-trace into CSF along its flow path, because of a local outside-in concentration gradient at the border of the subarachnoid space. Although lumbar CSF beta-trace concentrations may differ substantially from the concentrations of CSF beta-trace protein in the subarachnoid space of the optic nerve, our hypothesis could be tested by measuring beta-trace concentrations in lumbar CSF. Indeed, although the finding of higher CSF beta-trace concentrations in the subarachnoid space of the optic nerve in NTG patients could also be interpreted as evidence of impaired circulation of CSF in a compartmented subarachnoid space of the optic nerve, higher lumbar CSF beta-trace concentrations in NTG patients would add support to our hypothesis of CSF circulatory failure, given the reported increase of beta-trace concentration in lumbar CSF in case of decreasing CSF flow rate. Aside from that, the question of course is whether the use of lumbar puncture in patients with NTG could be ethically justified, given the relatively invasive nature of this procedure.
Interestingly, some anecdotal evidence for our hypothesis comes from a recent study in which a concentration gradient of beta-trace protein between the spinal CSF and the CSF in the subarachnoid space of the optic nerve was measured in 3 patients with idiopathic intracranial hypertension and in 2 patients with NTG. In contrast to the patients with elevated intracranial pressure, an inversion of the beta-trace protein ratio (beta-trace optic nerve / beta-trace lumbar) was measured in the 2 patients with NTG. Indeed, in the 2 cases of NTG, the beta-trace concentration was higher in the lumbar CSF than in the CSF from the subarachnoid space of the optic nerve. Reference values for beta-trace protein in normal human CSF are 15.3 ± 2.7 mg/L. Remarkably, compared with these normal values, the lumbar CSF beta-trace concentrations were significantly elevated in the 2 patients with NTG (34.10 mg/L and 73.20 mg/L in the oldest patient with NTG). Although this anecdotal evidence is encouraging, it should be interpreted with considerable caution, given the small number of patients with NTG.
Seemingly inconsistent with the present hypothesis, in their retrospective study of CSF pressure measurement in patients with glaucoma, Berdahl and associates also found that nonspecific protein concentrations in CSF were significantly lower in the POAG group (58.1 ± 23.0 mg/dL vs 68.1 ± 28.2 mg/dL, P < .05) and the NTG subset (45.5 ± 22.6 mg/dL vs 68.1 ± 28.2 mg/dL, P = .01) compared with that in the control subjects. The authors noted that their retrospective design did not allow further pursuit of this finding, but suggested the possibility that optic nerve damage might lead to a reduction in specific proteins or that a deficiency in specific proteins within the CSF might lead to or augment glaucomatous damage. Even though the NTG sample number was low, with only 11 subjects, this group had the lowest CSF protein concentration. At first sight, this finding seems to argue against our idea that reduced CSF production may contribute to low CSF pressure in NTG patients. Indeed, a decrease of CSF flow may lead to an increased CSF protein concentration. However, these CSF protein concentration data should be interpreted with caution. Indeed, CSF protein concentrations can be influenced by many factors other than CSF formation rate and turnover. For example, many neurologic diseases are accompanied by increased protein concentrations in the CSF. The population studied by Berdahl and associates included patients with neurologic signs and symptoms serious enough to warrant lumbar puncture, which can bias the results. The reasons for lumbar puncture were substantially different between the NTG and control groups. In the NTG group, the indications for lumbar puncture were altered mental status (64%), headache (18%), and stroke (9%). In the control group, the indications for lumbar puncture were altered mental status (26%), headache (27%), meningitis (12%), normal pressure hydrocephalus (5%), seizure (8%), stroke (5%), peripheral neuropathy (9%), radiculopathy (6%), and carcinomatosis (2%). Because many neurologic diseases may influence the concentration of CSF proteins, it is likely that some of the above neurologic diseases may have affected the values of CSF protein concentration in the NTG and control groups. Consequently, the difference in CSF protein concentration found between the 2 groups could result, at least in part, from differences in group composition.
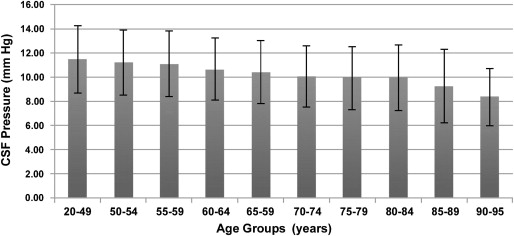
Interestingly, the present hypothesis is consistent with the age-specific pattern of prevalence of NTG. As noted above, the pathogenesis of NTG remains poorly understood. It is possible that NTG is a heterogeneous group of diseases as a result of various etiologies. Vascular dysregulation and blood flow disturbances have been reported, as NTG is often accompanied by a variety of cardiovascular and hematologic abnormalities. It should be stressed that CSF circulatory failure eventually accounts only for a subgroup of NTG patients. It is possible that CSF stagnation and subsequent accumulation of neurotoxins in the subarachnoid space surrounding the optic nerve may be correlated with NTG, especially in older patients, whereas younger NTG patients have glaucomatous damage independent of CSF circulatory dysfunction (eg, vasospastic phenomena are more often found in younger NTG patients). Although there is a substantial proportion of patients who are under the age of 50 years, with figures ranging from 11%-30% of all cases, NTG is considered to be a disease of elderly people. In the Beaver Dam Eye Study, age-specific prevalence rates of NTG were as follows: 0.20% for those 43-54 years of age; 0.68% for those 55-64 years of age; 0.62% for those 65-74 years of age; 1.61% for those over 75 years of age. Given that a decreased CSF pressure can be the consequence of reduced CSF production and turnover, it is interesting to note that a very recent study demonstrated that CSF pressure decreases with older age. Fleischman and associates retrospectively examined the charts of 33 922 patients who had a lumbar puncture between 1996 and 2009. Of these, 12 118 patients met all entry criteria. Mean CSF pressure within all age groups is shown in the figure . Relative to mean CSF pressure at age group 20-49 (mean 11.5 ± 2.8 mm Hg), mean CSF pressure declined steadily after age 50, with percent reduction of 2.5% for the 50-54 age group (mean 11.2 ± 2.7 mm Hg; P < .002) to 26.9% for the 90-95 age group (mean 8.4 ± 2.4 mm Hg; P < .001). Interestingly, the age where CSF pressure begins to decline coincides with the age where the prevalence of NTG increases.