Fig. 6.1
Retinoblastoma arises during retinal development. (a) The mature retina is made up of seven major classes of retinal cell types (rods, cones, ganglion cells, bipolar cells, horizontal neurons, amacrine cells, and Müller glia). (b) During development, multipotent retinal progenitor cells produce each of the retinal cell types in an evolutionary conserved birth order. Retinal birth order is overlapping with horizontal cells, cones, and ganglion cells born early during development and rods, bipolars, and Müller glia born at the end of retinogenesis. Retinoblastoma arises in the developing retina as progenitor cells produce retinal neurons. It is not known which cell type gives rise to retinoblastoma, but the tumors have a hybrid differentiation signature of progenitor cells, rods, cones, amacrine, and horizontal neurons
The evidence for a retinal progenitor cell as the retinoblastoma cell of origin comes from several experiments using genetic studies in mice. First, conditional inactivation of Rb and p107 in newly postmitotic rod photoreceptors (~80 % of the cells in the mouse retina) did not result in retinoblastoma [23], but when RB1 and p107 were inactivated in proliferating retinal progenitor cells using three different independent approaches, retinoblastomas developed in all three models [13–15]. Further evidence that a retinal progenitor cell can give rise to retinoblastoma in mice came from studies using replication-incompetent retroviral vectors. Expression in the retina of the adenoviral E1A oncogene, which inhibits Rb family members, using a retroviral vector that can only infect proliferating cells caused deregulated proliferation in retinal progenitor cells [12, 13]. Individual infected retinal progenitor cells expressing the E1A oncogene formed clonal focal retinal hyperplastic lesions, and simultaneous elimination of p53 led to formation of frank retinoblastomas [12, 13].
One approach that is often used to identify cancer cell of origin is to analyze differentiation markers. The main assumption of this approach is that the normal cell type that expresses a given protein may be the cell of origin for a cancer in which that protein is expressed. Retinoblastomas have been shown to express photoreceptor-specific genes, which initially suggested this cell type as the cell of origin [24]. However, further analysis has shown that human retinoblastoma samples express a variety of other cell-specific markers [21]. Indeed, a more recent comparison of gene expression array data of human and mouse retinoblastomas suggest that retinoblastomas co-express multiple differentiation pathways that are normally incompatible during retinogenesis [25]. These indeterminate results reflect the difficulty in the differentiation marker approach to cell of origin studies; tumor cells that express different markers could have arisen from different cell types, or they may simply have arisen from the same multipotent progenitor cell at a different point in maturation. Further, gene expression changes in retinoblastoma, which is a developmental regulator, may reflect a nonspecific, deregulated developmental program initiated by the loss of Rb. In mice, the picture is more straightforward. Retinoblastomas from knockout mice described above express markers of retinal progenitor cells and amacrine cells, and recent results with electron microscopy also are consistent with these cell types [25]. However, these results must be interpreted cautiously since even the knockout mouse models are not genetically identical to human retinoblastoma.
The evidence for a newly postmitotic cell as the retinoblastoma cell of origin also relies upon marker expression. The expression of amacrine cell markers in mouse retinoblastomas may indicate that a newly postmitotic cell committed to the amacrine cell fate is the cell that is susceptible to loss of Rb. The strongest evidence for a postmitotic cell of origin comes from the position of ectopically dividing cells in the apical-basal organization of the developing retina [22]. Normally, DNA synthesis (S phase) occurs in retinal precursor cells on the basal surface of the retina, M phase occurs on the apical surface, and the G1 and G2 phases occur during the transition from apical to basal surface (discussed in [26]). The fact that Rb/p107-deficient retinas exhibit an absence of additional mitoses at the apical surface and an increase in S phase cells where differentiation normally occurs suggests that newly postmitotic cells can give rise to retinoblastoma [22]. These results must be interpreted with caution in light of the fact that the genetic manipulations in these mice result in widespread disruption of normal retinal lamination, perturbing the normal position of retinal progenitor cells and newly postmitotic cells within the developing retina.
While we have narrowed our focus on retinal progenitor cells and newly postmitotic cells as the possible retinoblastoma cell of origin, additional studies are required to definitively determine which cell type(s) require Rb to avoid cell cycle deregulation and malignant transformation.
6.4 Events in Retinoblastoma Progression
While the initiating genetic event in retinoblastoma – biallelic inactivation of the RB1 gene – is well established, little is known about subsequent genetic events that contribute to retinoblastoma formation and progression. A major unexplained question is why loss of Rb does not trigger an apoptotic response that eliminates nascent retinoblastoma cells before clonal expansion can occur. Thus, there are several potential explanations for how retinoblastomas circumvent apoptosis: (1) there could be a heretofore unidentified member of the p53 pathway that functionally ablates the p53 response despite the presence of functional p53; (2) there may be genetic events in other apoptosis-related pathways, such as the Bcl2 pathway; or (3) the retinoblastoma cell of origin may be naturally resistant to apoptosis. In most cancers, there are mutations in the p53 tumor suppressor or other members of the p53 pathway that explain the acquired resistance to apoptosis [6]. Further, in mouse models of retinoblastoma, tumor development is greatly enhanced when p53 is inactivated [27]. However, there is no evidence that p53 is mutated frequently in human retinoblastomas [28]. Further, p53 can be activated in retinoblastomas, suggesting that the protein is functional [29]. Other members of the p53 pathway can be disrupted in cancer, leading to functional inhibition of p53. For example, the p53 inhibitor MDM2 is overexpressed in some forms of cancer [30]. Although there is no evidence to date for MDM2 alterations in human retinoblastoma, other genetic events have been identified, such as E2F3 amplification [31], which may disrupt the p53 response that is normally triggered by loss of Rb. Finally, retinoblastoma may represent a unique exception in which the p53 pathway is intact. There are several lines of evidence to support this possibility. First is the lack of apoptotic mutations described above. Second is the fact that human retinoblastomas typically have a very high rate of apoptosis, suggesting that the apoptotic response is still intact but that proliferation is simply outstripping apoptosis [32]. Third, there is recent evidence suggesting that the retinoblastoma cell of origin may be naturally resistant to apoptosis, which potentially assuages the need to postulate any genetic event subsequent to Rb inactivation that is necessary for retinoblastoma formation [14].
Over the past several years, there has been progress in identifying the pathways that may contribute to our understanding of how retinoblastomas overcome cell death. First, there is evidence that the p53 antagonist called MDM4 is upregulated in retinoblastoma and that there may be alternatively spliced oncogenic forms of MDM4 in retinoblastoma [25, 33]. Indeed, this is therapeutically relevant as the MDM2/MDM4 antagonist (nutlin-3a) can efficiently kill retinoblastoma cells [34]. Second, recent data suggest that the SYK oncogene is epigenetically upregulated in retinoblastoma and this promotes the stabilization of the BCL2 family member called MCL1 (Figs. 6.2 and 6.3). Both SYK and MCL1 are required for retinoblastoma survival, and small-molecule inhibitors of SYK are effective at inducing retinoblastoma cell death in vitro and in vivo [35].
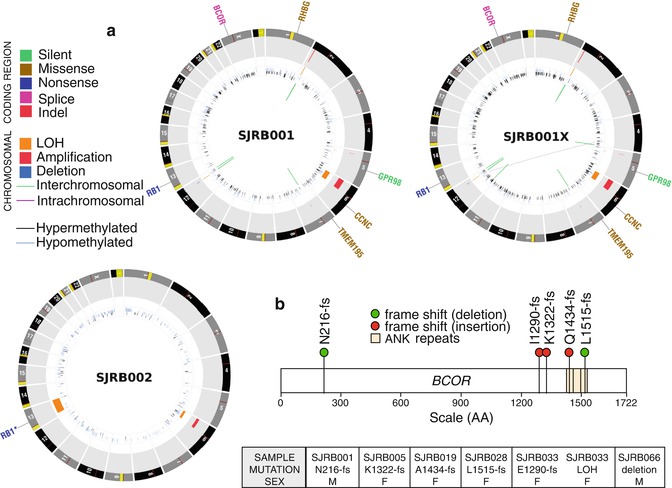
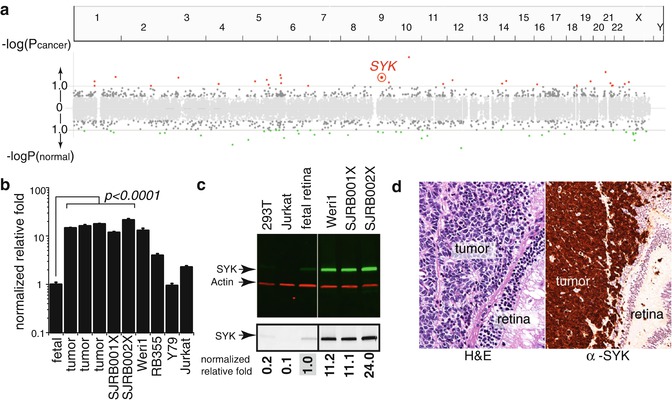
Fig. 6.2
Whole-genome sequencing of retinoblastoma. (a) CIRCOS plots of the whole-genome sequence data for representative retinoblastoma primary tumors (SJRB001 and SJRB002) and an orthotopic xenograft derived from one of those tumors (SJRB001X). (b) Beyond RB1, the only recurrently mutated gene was BCOR, an epigenetic regulator
Fig. 6.3
Integrated epigenetic analysis of retinoblastoma. (a) Plot of the epigenetically deregulated genes in retinoblastoma. These data are integrated from ChIP-on-chip, gene expression, and DNA methylation results. The spleen tyrosine kinase gene (SYK) was epigenetically upregulated in human retinoblastoma. (b) Validation of increase mRNA expression for SYK and validation of upregulation of SYK protein in retinoblastoma (c). Immunohistochemistry of 82 human retinoblastomas (d) showed upregulation of SYK in 100 % of tumors
Recent whole-genome sequencing of retinoblastomas showed that some retinoblastomas have very few mutations or chromosomal alterations (Fig. 6.2) [35]. Moreover, the passage of orthotopic xenografts in the eyes of immunocompromised mice retains stable genomes [35]. Taken together, these data suggest that genomic instability and the subsequent genetic lesions that may result from such instability are not required for retinoblastoma progression [35]. While inactivation of the RB1 gene in retinoblastoma may result in defects in sister chromatid cohesion [36], this does not necessarily lead to genomic instability. Instead, it has been shown that inactivation of the RB1 gene leads to massive epigenetic deregulation of known oncogenes and tumor suppressors, and this may contribute to the rapid progression of the disease following the second mutational event in RB1 [35].
6.5 Retinoblastoma Without RB1 Mutations
Recently, Brenda Gallie proposed that a small subset of retinoblastomas (~1 %) may arise as a result of a single oncogenic event rather than biallelic inactivation of RB1. Specifically, they propose that some retinoblastomas have MYCN amplification and do not have any RB1 mutations [37]. While it is a very small number of patients, it is an intriguing hypothesis that will require independent validation across a broader cohort of retinoblastoma samples. It will also be necessary to rule out other mechanisms of RB1 gene inactivation such as chromosomal events such as chromothripsis. This is important because chromothripsis at the RB1 locus would result in inactivation of the gene but would not be detected by conventional methods of RB1 gene analysis used by Gallie. Specifically, exon sequence analysis, promoter methylation analysis, analysis of LOH, and copy number changes would all appear to be wild type in a tumor where RB1 is inactivated by chromothripsis. All the exons of RB1 would be present, the promoter would be hypomethylated, both copies of RB1 would be present, and any copy number changes would be minimal. Thus, the tumor would appear to be wild type for RB1, but it would actually have a gene inactivation. Whole-genome sequencing combined with fluorescence in situ hybridization is currently the only way to identify retinoblastoma samples with chromothripsis. Future studies will be required to determine how many of the retinoblastomas with MYCN amplification actually have a wild-type RB1 gene and produce functional protein.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


