Retinoblastoma: Introduction, Genetics, and Clinical Features
Introduction
Retinoblastoma is the most common intraocular malignancy of childhood, occurring in about 1 in 15,000 live births. Numerous articles have been published about this important neoplasm, only a few of which are cited here (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42). Unlike uveal melanoma, it has no predilection for gender or race. It can be unilateral or bilateral, with the bilateral cases invariably representing germinal mutations. The overall mean age at diagnosis is 18 months. For unilateral cases, it is 24 months, and for bilateral cases, 12 months. However, it can be present at birth or have its onset in the teenage years or adulthood (7, 8, 9).
Genetics
The familial form of retinoblastoma has been associated with a deletion of the long arm of chromosome 13 (4, 5, 6). Affected patients are usually systemically normal but can sometimes have overt clinical signs of the 13q- syndrome.
Patients with the genetic mutation for retinoblastoma have a predisposition to a number of second tumors, particularly sarcomas, with osteosarcoma of long bones being the most frequent (10, 11, 12, 13, 14, 15, 16). The familial form of retinoblastoma is also associated with a higher incidence of pinealoblastoma and other parasellar tumors, neoplasms that are very similar to retinoblastoma from embryologic, anatomic, and immunologic standpoints (17, 18, 19, 20, 21, 22, 23). The association of bilateral retinoblastoma with pinealoblastoma has been termed trilateral retinoblastoma. This term can be inaccurate, in that some patients with the gene deletion and pinealoblastoma have unilateral retinoblastoma or no retinoblastoma.
Clinical Features
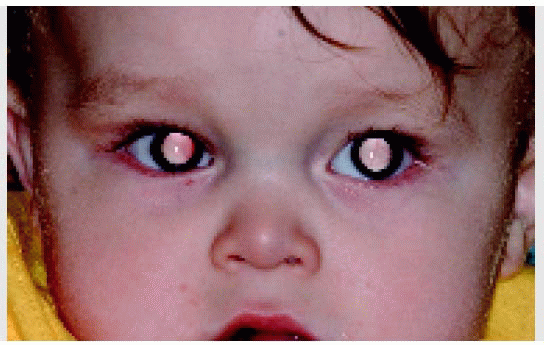
Clinically, retinoblastoma begins as a small, transparent lesion in the sensory retina that may be easily overlooked with ophthalmoscopy. As the tumor enlarges, it becomes opaque white and develops a dilated retinal feeding artery and draining vein, and a secondary retinal detachment can occur. The best-known initial clinical sign is a white pupillary reflex, called leukocoria (1, 2, 3). As the tumor enlarges, it can leave its intraretinal location and assume an exophytic growth pattern, endophytic growth pattern, or a combination of the two. The exophytic pattern is characterized by growth of the tumor outward toward the subretinal space, producing an overlying retinal detachment. The endophytic pattern is characterized by seeding of tumor cells into the overlying vitreous, obscuring a clear view of the retina.
The less common diffuse growth pattern is characterized by flat growth of the tumor (24, 25, 26, 27); although diffuse retinoblastoma is usually in the posterior retina, it has rarely appeared near the ora serrata and over the ciliary body without posterior involvement (28). Retinoblastoma can produce secondary glaucoma in about 17% of cases, usually due to iris neovascularization and secondary angle closure (29). The iris neovascularization can lead to a spontaneous hyphema, a rare presenting sign (25,32). Inflammation from necrotic intraocular retinoblastoma can simulate or cause orbital cellulitis (30). Although the lens is usually clear and in the correct position, there is a rare association with cataract and subluxed lens. It can extend extrasclerally and present as an advanced fungating mass.
Spontaneous Regression
Retinoblastoma has a tendency to undergo spontaneous regression in about 5% of cases. Spontaneously regressed retinoblastoma was initially recognized early to have rather typical clinical features. Subsequently, a benign variant of retinoblastoma, called retinoma or retinocytoma, was described. We prefer to use the term spontaneously regressed retinoblastoma for a tumor that actually grows to a certain size and then regresses. We use the term spontaneously arrested retinoblastoma for the benign variant (retinoma; retinocytoma) that grows to a certain size and then stabilizes (1). It is most likely that some cases reported as spontaneously regressed retinoblastoma represent these spontaneously arrested tumors.
International Classification of Retinoblastoma
An International Classification of Retinoblastoma has recently been proposed and is expected to be used at all major centers where retinoblastoma patients are managed. The details of this classification and its implications for predicting prognosis for ocular salvage using chemoreduction and supplemental treatment have been reported (42). The classification is summarized in the following table.
International Classification of Retinoblastoma (ICRB)
3. McLean IW. Retinoblastomas, retinocytomas, and pseudoretinoblastomas. In: Spencer WH, ed. Ophthalmic Pathology. An Atlas and Textbook, 4th ed. Philadelphia: WB Saunders; 1996:1340-1375.
4. Sparkes RS, Murphree AL, Lingua RW, et al. Gene for hereditary retinoblastoma assigned to human chromosome 13 by linkage to esterase D. Science 1983;219:971-973.
5. Shields CL, Shields JA, Donoso LA. Clinical genetics of retinoblastoma. In: Shields JA, ed. Update on Malignant Ocular Tumors. International Ophthalmology Clinics. Boston: Little, Brown; 1993;33:67-76.
8. Shields JA, Michelson JB, Leonard BC, et al. Retinoblastoma in an 18-year-old male. J Pediatr Ophthalmol 1976;13:275-277.
9. Shields CL, Shields JA, Shah P. Retinoblastoma in older children. Ophthalmology 1991;98:395-399.
10. Abramson DH, Ellsworth RM, Zimmerman LE. Nonocular cancer in retinoblastoma survivors. Trans Am Acad Ophthalmol 1976;81:454-456.
11. Roarty JD, McLean IW, Zimmerman LE. Incidence of second neoplasms in patients with bilateral retinoblastoma. Ophthalmology 1988;95:1583-1587.
12. Moll AC, Imhof SM, Bouter LM, et al. Second primary tumors in patients with hereditary retinoblastoma: a register-based follow-up study, 1945-1994. Int J Cancer 1996;67:515-519.
13. Abramson DH, Frank CM. Second nonocular tumors in survivors of bilateral retinoblastoma: a possible age effect on radiation-related risk. Ophthalmology 1998;105:573-579.
14. Shields JA, Husson M, Shields CL, et al. Orbital malignant fibrous histiocytoma following irradiation for retinoblastoma. Ophthalmic Plast Reconstr Surg 2001;17:58-61.
15. Rundle P, Shields JA, Shields CL, et al. Sebaceous gland carcinoma of the eyelid 16 years after irradiation for retinoblastoma. Eye 1999;13:109-110.
16. Moll AC, Imhof SM, Schouten-Van Meeteren AY, et al. Second primary tumors in hereditary retinoblastoma: a register-based study, 1945-1997: is there an age effect on radiation-related risk? Ophthalmology 2001;108:1109-1114.
17. Bader JL, Meadows AT, Zimmerman LE, et al. Bilateral retinoblastoma with ectopic intracranial retinoblastoma: trilateral retinoblastoma. Cancer Genet Cytogenet 1982;5:203-213.
18. Donoso LA, Shields JA, Felberg NT, et al. Intracranial malignancy in patients with bilateral retinoblastoma. Retina 1981;1:67-74.
19. Kivela T. Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol 1999;17:1829-1837.
20. Pesin SR, Shields JA. Seven cases of trilateral retinoblastoma. Am J Ophthalmol 1989;107:121-126.
21. Singh AD, Shields CL, Shields JA. New insights into trilateral retinoblastoma. Cancer 1999;86:3-5.
22. Marcus DM, Brooks SE, Leff G, et al. Trilateral retinoblastoma: insights into histogenesis and management. Surv Ophthalmol 1998;43:59-70.
23. De Potter P, Shields CL, Shields JA. Clinical variations of trilateral retinoblastoma. Areport of 13 cases. J Pediatr Ophthalmol Strabismus 1994;31:26-31.
24. Nicholson DH, Norton EW. Diffuse infiltrating retinoblastoma. Trans Am Ophthalmol Soc 1980;78:265-289.
25. Shields JA, Shields CL, Materin M. Diffuse infiltrating retinoblastoma presenting as a spontaneous hyphema. J Pediatr Ophthalmol Strabismus 2000;37:311-312.
26. Shields JA, Shields CL, Eagle RC, et al. Spontaneous pseudohypopyon secondary to diffuse infiltrating retinoblastoma. Arch Ophthalmol 1988;106: 1301-1302.
Only gold members can continue reading. Log In or Register to continue
 Retinoblastoma: Introduction, Genetics, Clinical Features, Classification
Retinoblastoma: Introduction, Genetics, Clinical Features, Classification


