 Tumors and Related Lesions of the Pigment Epithelium
Tumors and Related Lesions of the Pigment EpitheliumSolitary Congenital Hypertrophy of the Retinal Pigment Epithelium
General Considerations
Tumors and related lesions of the retinal pigment epithelium (RPE) include congenital hypertrophy of the RPE (CHRPE), hyperplastic RPE lesions associated with familial adenomatous polyposis, congenital simple hamartoma, combined hamartoma, pseudocancerous reactive hyperplasia, benign epithelioma (adenoma), and malignant epithelioma (adenocarcinoma). CHRPE is further divided into solitary and multifocal variants, the latter also known as congenital grouped pigmentation or “bear tracks” (1).
Solitary CHRPE is a well-known fundus condition that has been the subject of considerable attention in the ophthalmic literature (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22). Although it is believed to be congenital, the median age at diagnosis is 45 years (2). This late diagnosis may be because the condition is generally asymptomatic and located in midperipheral or peripheral fundus and may remain undetected on routine fundus examination. It is usually a solitary lesion that, unlike choroidal nevus and melanoma, has no recognized predilection for race.
The term CHRPE has also been applied to a somewhat different multifocal fundus condition that has a high association with familial adenomatous polyposis (FAP) and bowel cancer (1,21), a subject to be discussed later. That choice of terminology is unfortunate because the typical solitary CHRPE apparently is not associated with an increased incidence of FAP or gastrointestinal malignancy (8).
Clinical Features
A review of 330 cases of solitary CHRPE provided additional information regarding this condition (2). It appears clinically as a well-demarcated flat to minimally elevated fundus plaque that can range from a black homogeneous lesion to a completely depigmented lesion (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16). As mentioned earlier, most are located in the midperipheral or peripheral fundus. Only 2% are found in the macular or peripapillary region. The lesion is predominately pigmented in 88% and nonpigmented in 12% of cases. The median largest basal diameter is 4.5 mm. Well-defined depigmented foci, called lacunae, are present in 43% of the pigmented CHRPE. Most solitary CHRPE lesions have a typical depigmented ring or “halo” around the margin. The lacunae show enlargement in 32% of cases after a mean follow-up of 5 years (2). The majority of solitary CHRPE show slightly increased diameter, as observed in 74% to 83% of cases followed >5 years in two reported series (2,6).
Concerning differential diagnosis, the large lesions, particularly those in the peripheral fundus, can appear similar to choroidal melanoma. The peripheral lesions generally give the false impression ophthalmoscopically that they are quite elevated. In a series of 330 cases, the referral diagnosis was choroidal nevus in 26%, choroidal melanoma in 15%, and nonspecified fundus lesions in 48%. The correct diagnosis of CHRPE was submitted in only 9% of cases. The clinical features described here are different from those of nevus or melanoma, and the diagnosis readily can be made by the experienced observer.
Recent reports have provided further information about the potential of solitary CHRPE to spawn a nodular growth pattern (9). In perhaps 2% of cases, such a nodular growth develops in the area of CHRPE (2). The growth the nodular growth gradually acquires a retinal feeding artery and draining vein and produces yellow intraretinal exudation and exudative retinal detachment (10). This pattern is identical to the adenoma of the RPE to be described subsequently. In one case that was managed by eye wall resection, the growing amelanotic nodular lesion arising from solitary CHRPE proved to be a low-grade malignant epithelioma (adenocarcinoma) of the RPE (10). A similar case was subsequently reported (15). The subject is discussed later under the section on epitheliomas of the RPE.
Diagnostic Approaches
Fluorescein angiography and indocyanine green angiography generally show blockage of fluorescence throughout most of the sequence. Characteristically, there is persistent hypofluorescence of the pigmented areas and transmission of choroidal fluorescence through the depigmented areas throughout the angiography sequence (1,2,4). Ultrasonography is nondiagnostic, but in many cases, it shows the lesion to be about 0.5 to 1 mm thick. Visual field results range from a mild relative scotoma to an absolute field defect, usually depending on the size of the lesion. Optical coherence tomography shows overlying retinal thinning, loss of photoreceptors, and moderate relative shadowing of the underlying choroid, but the findings are nondiagnostic (19). The lesion cannot generally be detected with CT or MRI.
Pathology
Histopathologically, the RPE cells in CHRPE are taller and more densely packed with spherical melanosomes as compared to the normal RPE, which has smaller, more elliptically shaped melanosomes. There appears to be a combination of cellular hyperplasia and hypertrophy. There is overlying atrophy of the photoreceptors (5).
Management
The management of solitary CHRPE is simple periodic observation. If a small nodular growth should evolve, it can often be observed for a period of time because progression is very slow and does not usually affect the patient’s vision. If such a nodule produces exudation or subretinal fluid, laser photocoagulation or cryotherapy can be considered to stop the progression of the leakage. If such a growth should produce surface wrinkling retinopathy in the macular region, vitrectomy and membrane peeling should be considered. The prognosis for CHRPE is generally excellent.
Selected References
1. Shields JA, Shields CL. Tumors and related lesions of the pigment epithelium. In: Shields JA, Shields CL, eds. Intraocular Tumors. A Text and Atlas. Philadelphia: WB Saunders; 1992:43-60.
2. Shields CL, Mashayekhi A, Ho T, et al. Solitary congenital hypertrophy of the retinal pigment epithelium: clinical features and frequency of enlargement in 330 patients. Ophthalmology 2003;110:1968-1976.
3. Gass JDM. Focal congenital anomalies of the retinal pigment epithelium. Eye 1989;3:1-18.
4. Purcell JJ, Shields JA. Hypertrophy with hyperpigmentation of the retinal pigment epithelium. Arch Ophthalmol 1975;93:1122-1126.
5. Lloyd WC III, Eagle RC, Shields JA, et al. Congenital hypertrophy of the retinal pigment epithelium: electron microscopic and morphometric observations. Ophthalmology 1990;97:1052-1060.
6. Chamot L, Zografos L, Klainguti G. Fundus changes associated with congenital hypertrophy of the retinal pigment epithelium. Am J Ophthalmol 1993;115:154-161.
7. Traboulsi EI, Maumenee IH, Krush AJ, et al. Pigmented ocular fundus lesions in the inherited gastrointestinal polyposis syndromes and in hereditary nonpolyposis colorectal cancer. Ophthalmology 1988;95:964-969.
8. Shields JA, Shields CL, Shah P, et al. Lack of association between typical congenital hypertrophy of the retinal pigment epithelium and Gardner’s syndrome. Ophthalmology 1992;99:1705-1713.
9. Shields JA, Shields CL, Singh AD. Acquired tumors arising from congenital hypertrophy of the retinal pigment epithelium. Arch Ophthalmol 2000; 118:637-641.
10. Shields JA, Shields CL, Eagle RC Jr, et al. Adenocarcinoma arising from congenital hypertrophy of retinal pigment epithelium. Arch Ophthalmol 2001;119:597-602.
11. Paoli D. Retinal astrocytoma associated with hypertrophy of the retinal pigment epithelium: clinical report and follow-up. Ophthalmologica 1998;212:71-73.
12. van der Torren K, Luyten GP Progression of papillomacular congenital hypertrophy of the retinal pigment epithelium associated with impaired visual function. Arch Ophthalmol 1998;116:256-257.
13. Sharma MC, Blake CR, Weinstein R, et al. Peripapillary congenital hypertrophy of the retinal pigment epithelium. Ophthalmic Surg Lasers Imaging 2004;35:174-175.
14. Shields JA. Tumors and related lesions of the retinal pigment epithelium. In: Guyer DR, Yannuzzi LA, Chang S, et al., eds. Retina-Vitreous-Macula, Vol. 2. Philadelphia: WB Saunders; 1999:1116-1129.
15. Trichopoulos N, Augsburger JJ, Schneider S. Adenocarcinoma arising from congenital hypertrophy of the retinal pigment epithelium. Graefes Arch Clin Exp Ophthalmol 2005;28:1-4.
16. Parsons MA, Rennie IG, Rundle PA, et al. Congenital hypertrophy of retinal pigment epithelium: a clinico-pathological case report. Br J Ophthalmol 2005;89:920-921.
17. Meyer CH, Rodrigues EB, Mennel S, et al. Grouped congenital hypertrophy of the retinal pigment epithelium follows developmental patterns of pigmentary mosaicism. Ophthalmology 2005;112:841-847.
18. Shields CL, Materin MA, Shields JA. Review of optical coherence tomography for intraocular tumors. Curr Opin Ophthalmol 2005;16:141-154.
19. Shields CL, Materin MA, Walker C, et al. Photoreceptor loss overlying congenital hypertrophy of the retinal pigment epithelium by optical coherence tomography. Ophthalmology 2006;113:661-665.
20. Traboulsi EI. Ocular manifestations of familial adenomatous polyposis (Gardner syndrome). Ophthalmol Clin North Am 2005;18:163-166.
21. Giuffre G. Indocyanine green angiography in congenital hypertrophy of the retinal pigment epithelium. Eur J Ophthalmol 2005;15:162-164.
22. van der Torren K, Luyten GP. Progression of papillomacular congenital hypertrophy of the retinal pigment epithelium associated with impaired visual function. Arch Ophthalmol 1998;116:256-257.
▪ Solitary Congenital Hypertrophy of the Retinal Pigment Epithelium (CHRPE): Clinical Variations
Solitary CHRPE can show considerable variation in color, size, and shape.
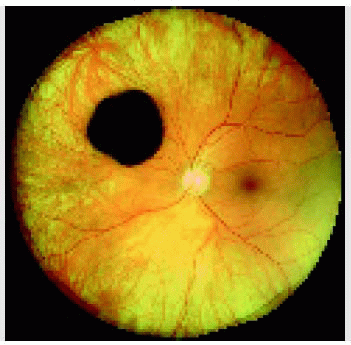
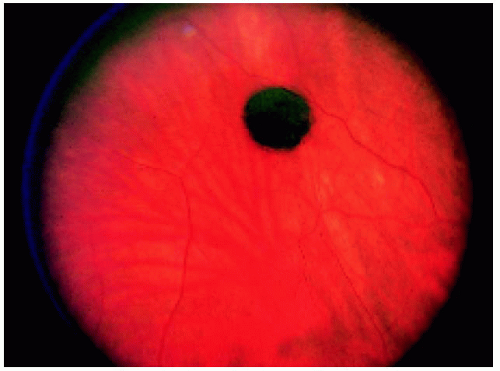 Figure 22.1. Small solitary congenital hypertrophy of the retinal pigment epithelium with a homogeneous black color in a 54-year-old man. |
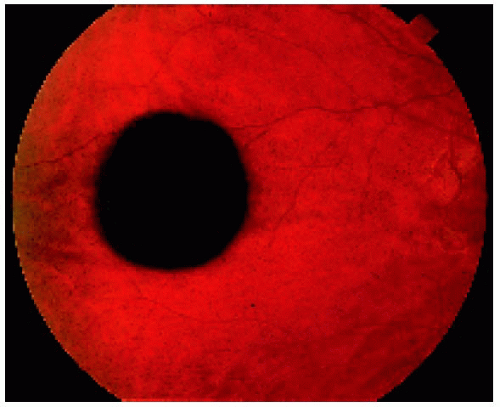 Figure 22.2. Slightly larger solitary congenital hypertrophy of the retinal pigment epithelium in a 17-year-old man. Note the black central portion and the slightly less pigmented peripheral area. |
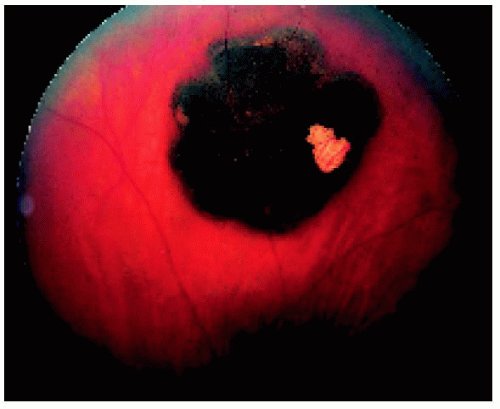 Figure 22.3. Solitary congenital hypertrophy of the retinal pigment epithelium, showing a depigmented lacuna and marginal light halo in a 28-year-old man. Note that the margin is slightly irregular but smooth. |
 Figure 22.4. Juxtapapillary solitary congenital hypertrophy of the retinal pigment epithelium (RPE) in a 40-year-old man, showing similar features to the more peripheral lesion shown in Figure 22.3. This is a very unusual location for congenital hypertrophy of the RPE. |
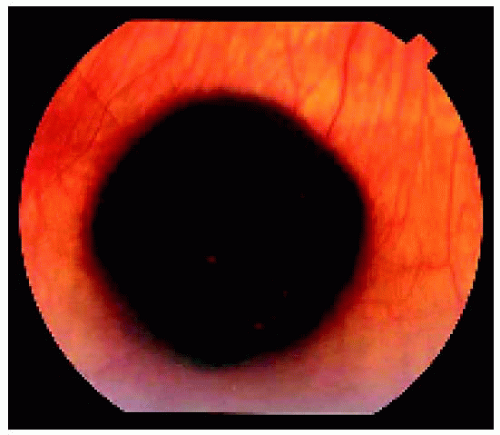 Figure 22.5. Solitary congenital hypertrophy of the retinal pigment epithelium located near the equator inferiorly in a middle-aged woman. Note the subtle lacunae in the lesion. |
 Figure 22.6. Large solitary congenital hypertrophy of the retinal pigment epithelium (RPE) in a 17-year-old man. He was referred for enucleation because of suspected choroidal melanoma, but the enucleation was canceled when the lesion was diagnosed as congenital hypertrophy of the RPE. |
▪ Solitary Congenital Hypertrophy of the Retinal Pigment Epithelium: Wide-Angle Imaging of Predominantly Pigmented Lesions
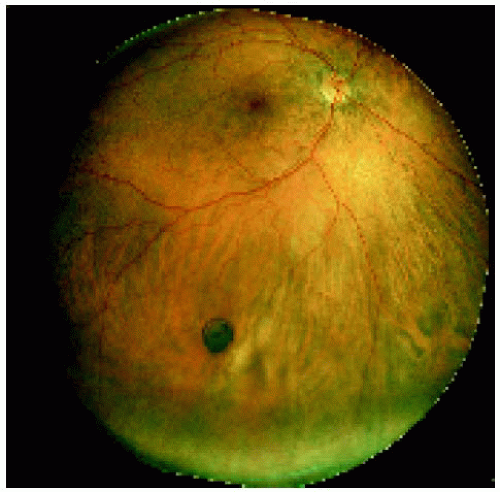 Figure 22.7. Small congenital hypertrophy of the retinal pigment epithelium located in the midperipheral fundus inferiorly. |
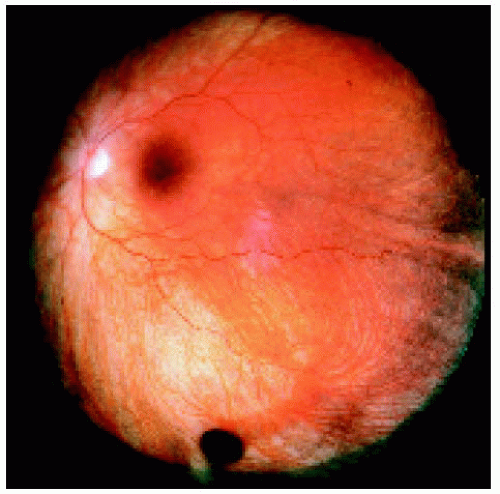 Figure 22.8. Small congenital hypertrophy of the retinal pigment epithelium located between the equator and the ora serrata inferiorly. |
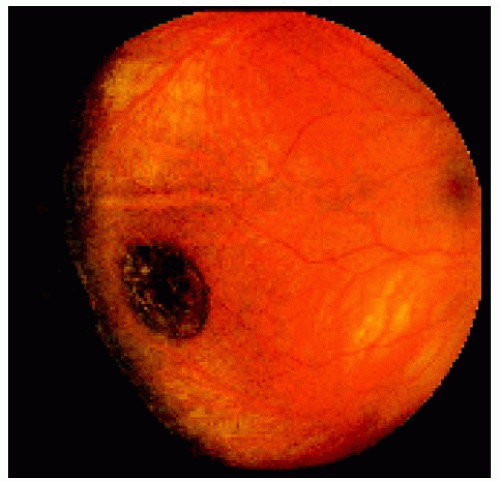 Figure 22.9. Medium-sized congenital hypertrophy of the retinal pigment epithelium located near the equator temporally. |
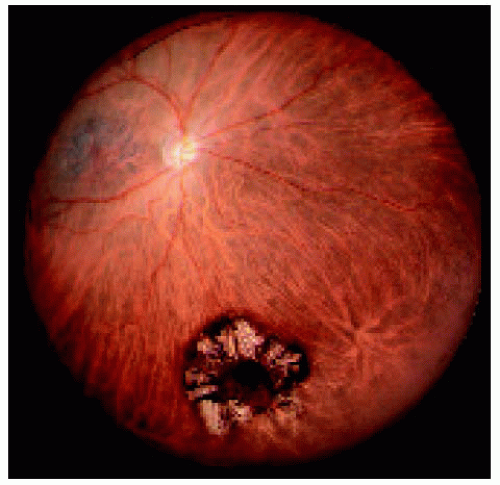 Figure 22.10. Medium-sized congenital hypertrophy of the retinal pigment epithelium (CHRPE) located near the equator inferiorly. Note the characteristic lacunae. There is a small pigmented nodule in the center of the lesion. The central nodule arising from CHRPE is discussed subsequently in the section on epithelioma of the RPE. |
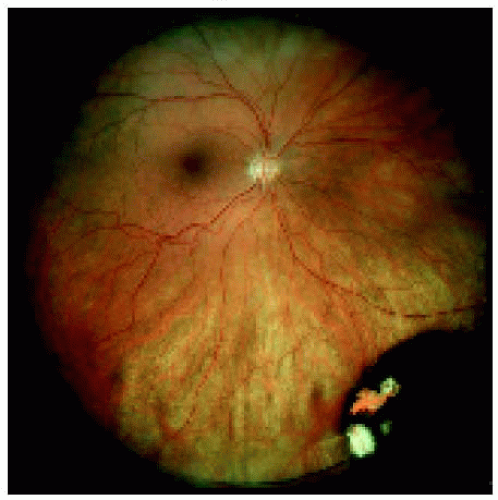 Figure 22.11. Medium-sized congenital hypertrophy of the retinal pigment epithelium located near the equator inferonasally. |
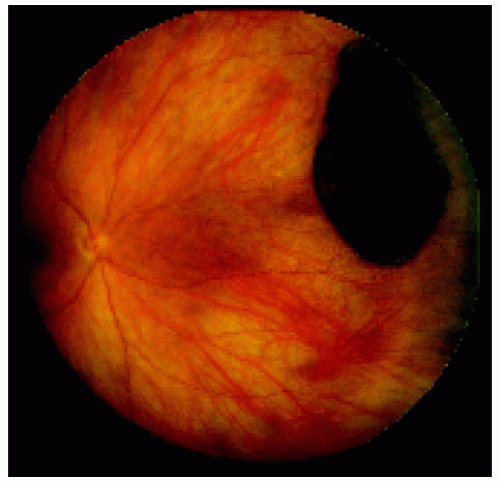 Figure 22.12. Large congenital hypertrophy of the retinal pigment epithelium located near the equator nasally. |
▪ Solitary Congenital Hypertrophy of the Retinal Pigment Epithelium: Wide-Angle Imaging of Predominantly Nonpigmented Lesions
In some instances, solitary congenital hypertrophy of the retinal pigment epithelium seems to lose its pigmented appearance and appear predominantly amelanotic. This may correspond with coalescence of the depigmented lacunae. Examples are shown.
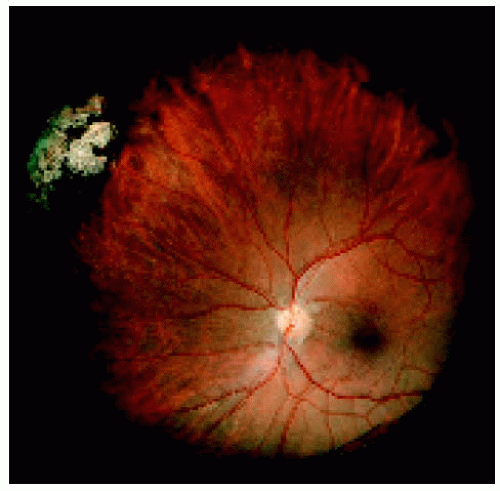 Figure 22.13. Medium-sized congenital hypertrophy of the retinal pigment epithelium located near the ora serrata superonasally. |
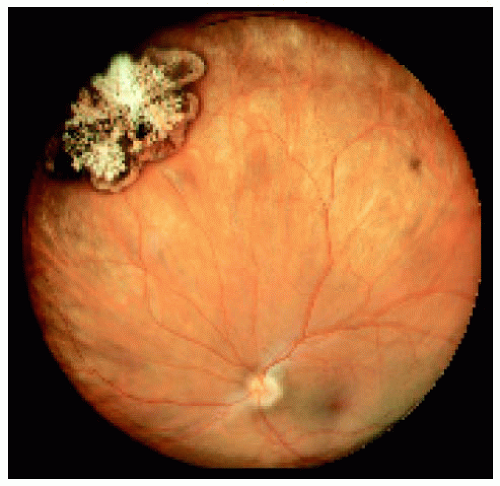 Figure 22.14. Medium-sized congenital hypertrophy of the retinal pigment epithelium located near the equator superonasally. |
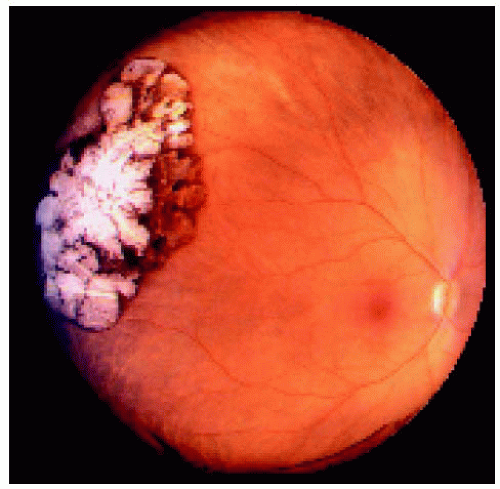 Figure 22.15. Large congenital hypertrophy of the retinal pigment epithelium located near the equator temporally. |
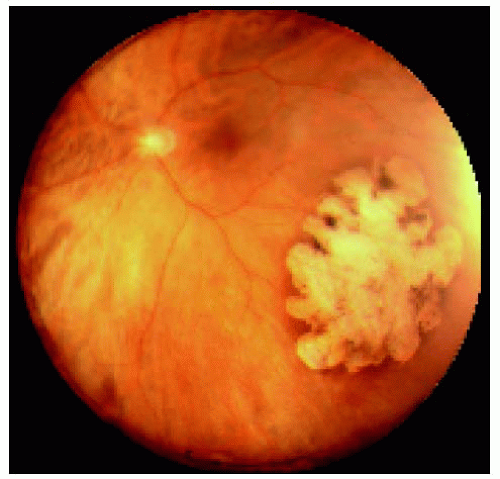 Figure 22.16. Large congenital hypertrophy of the retinal pigment epithelium located near the equator inferotemporally. |
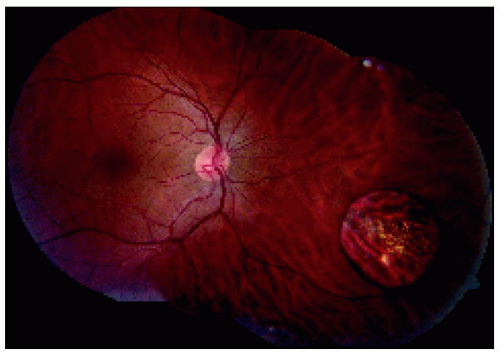 Figure 22.17. Montage showing medium-sized congenital hypertrophy of the retinal pigment epithelium located near the equator inferonasally. |
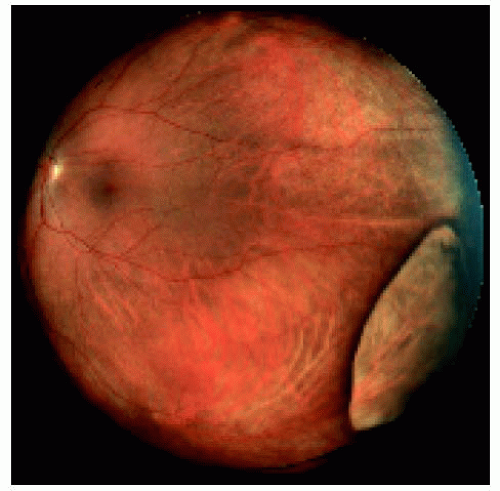 Figure 22.18. Large congenital hypertrophy of the retinal pigment epithelium located between the equator and the ora serrata inferotemporally. Note that the normal choroidal blood vessels can be seen through the lesion. |
▪ Solitary Congenital Hypertrophy of the Retinal Pigment Epithelium: Fluorescein Angiography and Histopathology
Lloyd WC III, Eagle RC, Shields JA, et al. Congenital hypertrophy of the retinal pigment epithelium: electron microscopic and morphometric observations. Ophthalmology 1990;97:1052-1060.
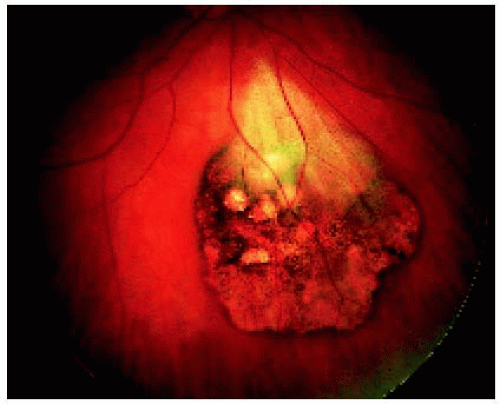 Figure 22.19. Solitary congenital hypertrophy of the retinal pigment epithelium, showing characteristic depigmented lacunae in a 46-year-old woman. The patch of myelinated retinal nerve fibers over the lesion is probably coincidental. |
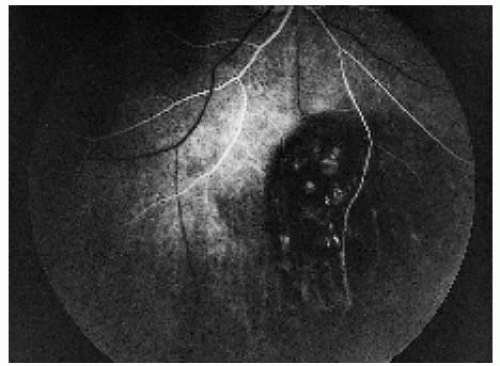 Figure 22.20. Arterial-phase fluorescein angiogram of the lesion shown in Figure 22.19, demonstrating early hypofluorescence of the lesion. |
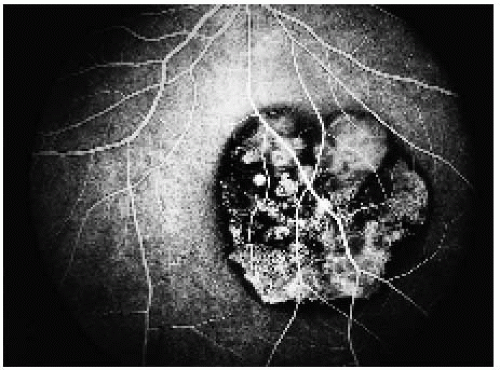 Figure 22.21. Full venous phase, showing hypofluorescence of the areas of pigmentation and transmission hyperfluorescence in the areas of depigmentation. |
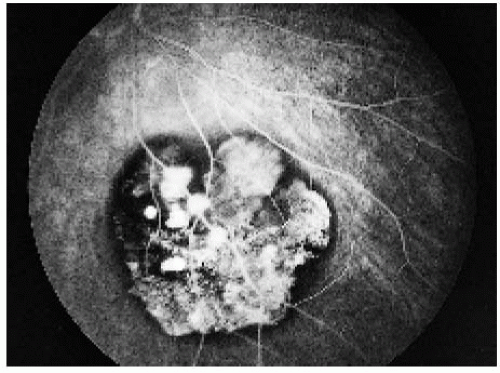 Figure 22.22. Late phase, showing a continued similar pattern of fluorescence. |
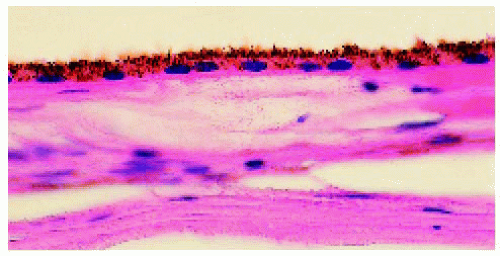 Figure 22.23. Pathology of normal retinal pigment epithelium (RPE) for comparison to congenital hypertrophy of the RPE. (Hematoxylin-eosin ×100.) |
 Figure 22.24. Pathology of congenital hypertrophy of the retinal pigment epithelium (RPE) from the same eye shown in Figure 22.23. Note that the RPE cells are slightly taller and more densely pigmented as compared to normal RPE cells. (Hematoxylin-eosin ×100.) |
▪ Solitary Congenital Hypertrophy of the Retinal Pigment Epithelium: Clinical and Optical Coherence Tomography Correlations
With optical coherence tomography, congenital hypertrophy of the retinal pigment epithelium typically shows retinal thinning and loss of photoreceptors in the overlying sensory retina. The more pigmented lesions tend to block light, and the less pigmented lesions tend to transmit light. Correlations are shown.
Shields CL, Materin MA, Walker C, et al. Photoreceptor loss overlying congenital hypertrophy of the retinal pigment epithelium by optical coherence. Ophthalmology 2006;113:661-665.
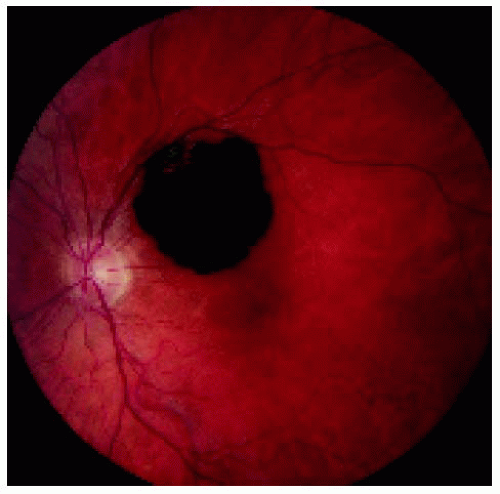 Figure 22.25. Solitary congenital hypertrophy of the retinal pigment epithelium superotemporal to the optic disc. |
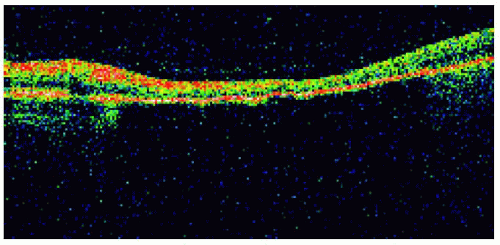 Figure 22.26. Optical coherence tomography of the lesion shown in Figure 22.25, demonstrating retinal thinning, loss of photoreceptors, and choroidal shadowing. |
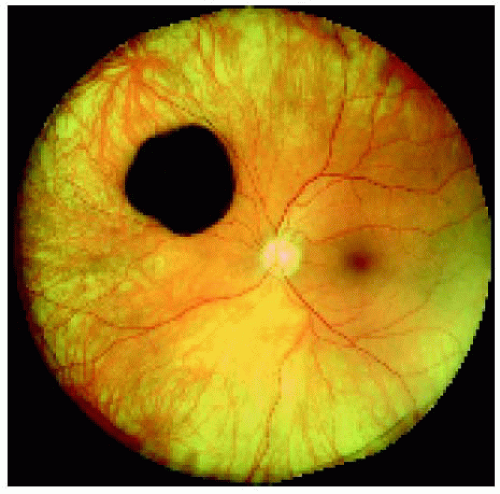 Figure 22.27. Solitary congenital hypertrophy of the retinal pigment epithelium superonasal to the optic disc. |
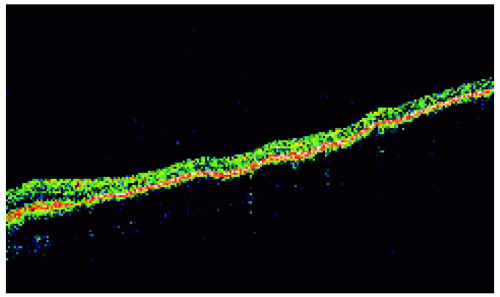 Figure 22.28. Optical coherence tomography of the lesion shown in Figure 22.27, revealing findings similar to the prior case. |
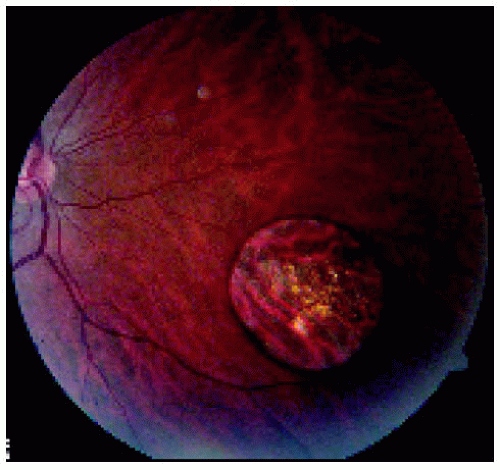 Figure 22.29. Solitary congenital hypertrophy of the retinal pigment epithelium inferonasal to the optic disc. |
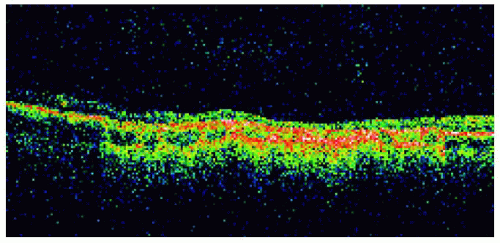 Figure 22.30. Optical coherence tomography of the lesion shown in Figure 22.29. |
▪ Solitary Congenital Hypertrophy of the Retinal Pigment Epithelium: Documented Growth in the Basal Dimension
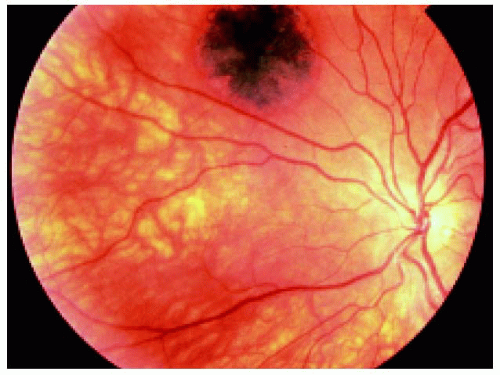 Figure 22.31. Small lesion superotemporal to the optic disc in the right eye of a 17-year-old woman. |
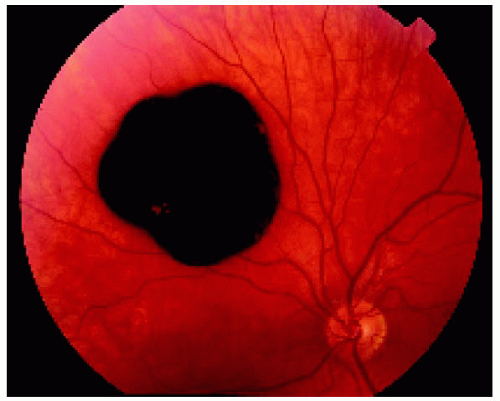 Figure 22.32. Appearance of the same lesion 13 years later, showing definite increase in its diameter. |
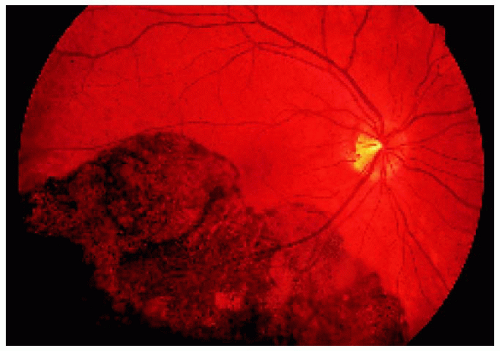 Figure 22.33. Large solitary congenital hypertrophy of the retinal pigment epithelium inferotemporal to the optic disc. |
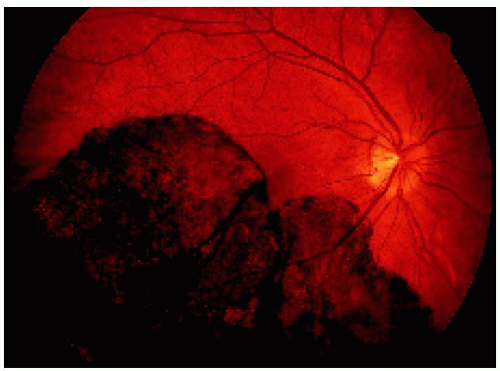 Figure 22.34. Appearance of the same lesion shown in Figure 22.33 after 2 years. Note that the lesion has enlarged slightly and is closer to the optic disc margin and now crosses the horizontal blood vessel that was not involved in the earlier photograph. |
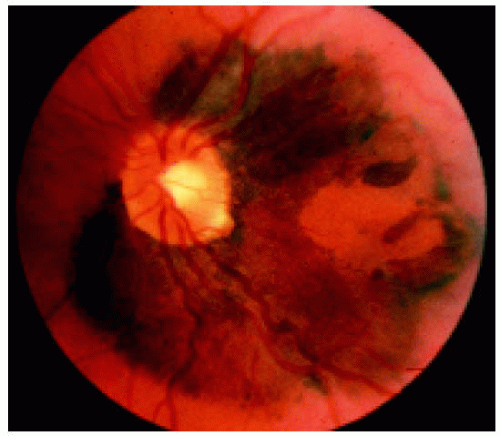 Figure 22.35. Peripapillary solitary congenital hypertrophy of the retinal pigment epithelium involving the macular region. (Courtesy of Gregg Luyten, MD.) |
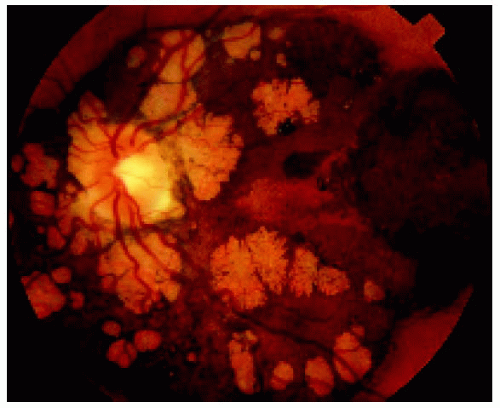 Figure 22.36. Lesion shown in Figure 22.35, several years later, demonstrating growth in the basal dimension and now encircling the optic disc. Note the numerous lacunae that have developed. (Courtesy of Gregg Luyten, MD.) |
Multifocal Congenital Hypertrophy of the Retinal Pigment Epithelium (Congenital Grouped Pigmentation; Bear Tracks)
General Considerations
The multifocal variant of CHRPE is also known as congenital grouped pigmentation of the retina or “bear tracks” (1, 2, 3, 4, 5, 6, 7, 8, 9). It is generally a nonhereditary, sporadic condition, but it has been observed in a mother and daughter (7). There are usually no related ocular abnormalities, but it has been observed in the opposite eye of a patient with persistent hyperplastic primary vitreous (8), retinoblastoma (4), neurofibromatosis (1), and Coats disease (3). These findings are possibly coincidental.
Clinical Features
Multifocal CHRPE is characterized by several groups of well-delineated, flat, slate-gray lesions that usually assume a sector distribution in the fundus. Each group consists of from 3 to 30 individual pigmented lesions that vary from 0.1 to 3 mm in diameter, with the larger lesions located more peripherally. Each individual lesion is similar to unifocal congenital hypertrophy of the RPE, but is usually smaller than the unifocal type and does not show the lacunae or haloes of depigmentation. The lesions are sometimes clinically nonpigmented, in which case they have been called “polar bear tracks” (9). These typical clinical features should serve to differentiate this condition from the numerous other conditions that can be associated with multiple pigmented lesions in the fundus. Retinal electrophysiologic studies are normal in multifocal CHRPE (6). The lesions tend to remain stable.
As mentioned earlier, a close relationship has recently been recognized between multiple small pigmented fundus lesions and familial adenomatous polyposis and Gardner syndrome, conditions associated with familial colonic cancer. The fundus lesions have been called CHRPE, but they are actually different from multifocal CHRPE in that they are bilateral, have a more haphazard distribution, and often have more irregular or jagged borders. There appears to be no association with the typical forms of CHRPE described here and these cancer syndromes (5). This subject is discussed further in the next section.
Pathology
Management
Multifocal CHRPE requires no treatment and can be observed as part of routine ocular examination. The prognosis is excellent.
Selected References
1. Shields JA, Shields CL. Tumors and related lesions of the pigment epithelium. In: Shields JA, Shields CL, eds. Intraocular Tumors. A Text and Atlas. Philadelphia: WB Saunders; 1992:43-60.
2. Shields JA. Tumors and related lesions of the retinal pigment epithelium. In: Guyer DR, Yannuzzi LA, Chang S, et al. Retina-Vitreous-Macula, Vol. 2, Philadelphia: WB Saunders; 1999;1116-1129.
3. Shields JA, T’so MOM. Congenital group pigmentation of the retina. Histopathologic description and report of a case. Arch Ophthalmol 1975;92: 1153-1155.
4. Regillo CD, Eagle RC Jr, Shields JA, et al. Histopathologic findings in congenital grouped pigmentation of the retina. Ophthalmology 1993;100: 400-405.
5. Shields JA, Shields CL, Shah P, et al. Lack of association between typical congenital hypertrophy of the retinal pigment epithelium and Gardner’s syndrome. Ophthalmology 1992; 99:1705-1713.
6. Yoshida T, Adachi-Usami E, Kimura T. Three cases of grouped pigmentation of the retina. Ophthalmologica 1995;209:101-105.
7. Renardel de Lavalette VW, Cruysberg JR, Deutman AF. Familial congenital grouped pigmentation of the retina. Am J Ophthalmol 1991;112:406-409.
8. Fujii M, Hayasaka S, Setogawa T. Persistent hyperplastic primary vitreous in the right eye and congenital grouped pigmentation of the retina in the left. Ophthalmologica 1989;198:135-139.
9. Gass JDM. Focal congenital anomalies of the retinal pigment epithelium. Eye 1989;3:1-18.
▪ Multifocal Congenital Hypertrophy of the Retinal Pigment Epithelium
Multifocal congenital hypertrophy of the retinal pigment epithelium is also known as congenital grouped pigmentation of the retina or “bear tracks.” Other than being multifocal, it is similar clinically and histopathologically to solitary CHRPE. Like the solitary form, it can be deeply pigmented or nonpigmented, but the depigmented variant is less common. It often assumes a sector distribution, with the small lesions located closer to the optic disc and large lesions toward the peripheral fundus.
Regillo CD, Eagle RC Jr, Shields JA, et al. Histopathologic findings in congenital grouped pigmentation of the retina. Ophthalmology 1993:100:400-405.
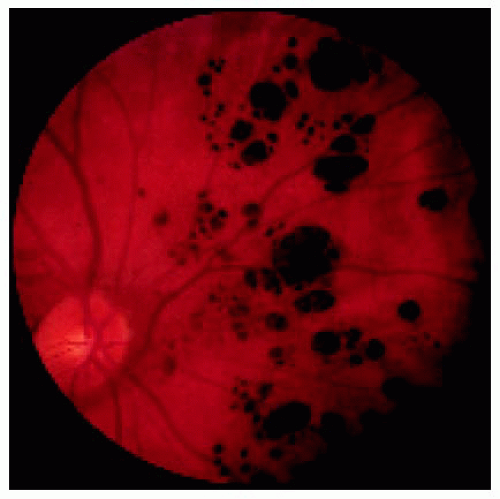 Figure 22.37. Typical multifocal congenital hypertrophy of the retinal pigment epithelium in a 1-year-old boy. |
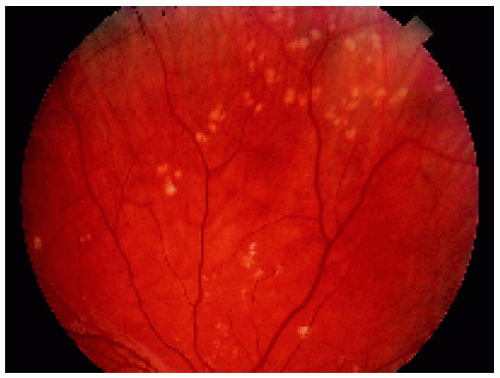 Figure 22.38. Nonpigmented congenital hypertrophy of the retinal pigment epithelium, sometimes called “polar beartracks,” in a 10-year-old girl. |
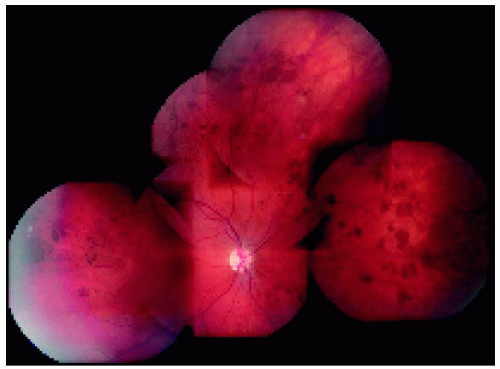 Figure 22.39. Widespread multifocal congenital hypertrophy of the retinal pigment epithelium in a young child. This child had very similar findings in the opposite eye. There was no family history of colon cancer. |
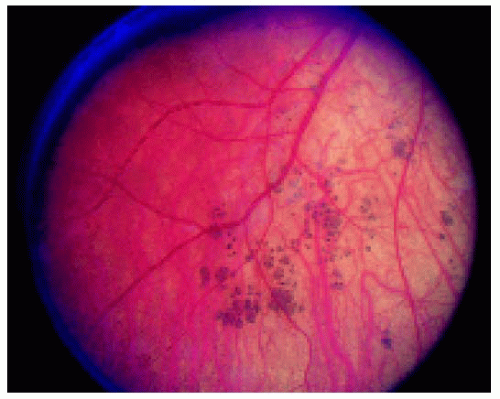 Figure 22.40. Multifocal congenital hypertrophy of the retinal pigment epithelium in a 2-year-old child with retinoblastoma. The opposite eye had similar lesions and was managed by enucleation, providing an opportunity to study the lesions pathologically. |
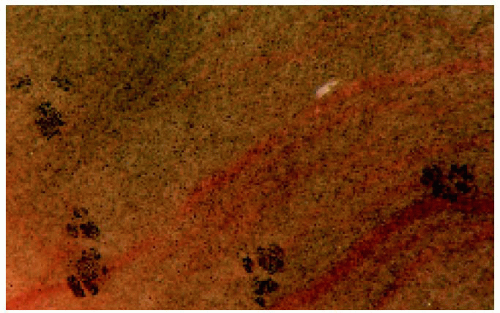 Figure 22.41. Gross photograph of the retinal pigment epithelium in the enucleated fellow eye of the patient seen in Figure 22.39, showing foci of hyperpigmentation. |
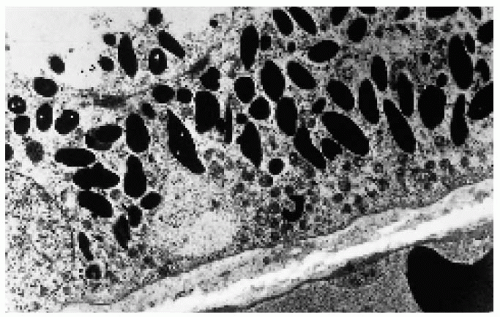 Figure 22.42. Electron microscopy of one of the lesions shown in Figure 22.41, demonstrating the large, densely packed melanosomes in the cytoplasm of the retinal pigment epithelium cells. (×4000.) (Courtesy of Ralph C. Eagle, Jr., MD.) |
Retinal Pigment Epithelial Hamartomas Associated with Familial Adenomatous Polyposis and Gardner Syndrome
General Considerations
This condition has a strong association with familial adenomatous polyposis, an autosomal dominant condition in which almost 100% of patients develop colon cancer (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23). It is unfortunate that the term CHRPE was used to describe this fundus condition by those who first made this association (3), because neither solitary nor multifocal CHRPE is associated with an increased incidence of colon cancer (4). To minimize confusion, we choose to call this condition “retinal pigment epithelial hamartomas associated with familial adenomatous polyposis” (RPEH-FAP). About 70% of patients with FAP have these characteristic fundus lesions, which represent the hallmark of a life-threatening familial cancer (14,17,18).
Many ophthalmologists have used the term Gardner syndrome to describe the association of these fundus lesions with bowel cancer. However, Gardner syndrome is defined as FAP plus extracolonic manifestations, including osteomas, desmoid tumors, cutaneous cysts, and several other tumors. Hence, all patients with Gardner syndrome have FAP, but patients with FAP do not necessarily have Gardner syndrome. There is also a close relationship between RPEH-FAP and central nervous system gliomas (Turcot syndrome) (6,16).
Clinical features
RPEH-FAP has characteristic ophthalmoscopic features. Unlike solitary CHRPE, the lesions are multiple and bilateral and their margins are often less well defined. Unlike multifocal CHRPE, they are randomly dispersed and do not have a sector distribution, and they have irregular depigmented margins, sometime forming a fish-tail or comma configuration (4,13). Some have proposed that the presence of three or more such lesions is diagnostic of FAP (18), but some patients have many more, even hundreds of lesions bilaterally.
Pathology and Pathogenesis
Histopathologically, RPEH-FAP has been identified to have one of three basic configurations: a monolayer of hypertrophic cells, a mound of RPE cells interposed between the RPE basement membrane of the inner collagenous layer of Bruch’s membrane, or a multilayered mound of hyperplastic cells (8). It is believed to indicate a generalized defect in melanogenesis. The gene for FAP has been identified on the long arm of chromosome 5 (5q21) (18).
Management
The management of RPEH-FAP is periodic observation only. However, the almost universal development of colon cancer in affected patients warrants close scrutiny for and early management of that malignancy. Affected patients, particularly those with a family history of colon cancer, should have periodic colonoscopy and removal of suspicious polyps.
Selected References
1. Shields JA, Shields CL. Tumors and related lesions of the pigment epithelium. In: Shields JA, Shields CL, eds. Intraocular Tumors. A Text and Atlas. Philadelphia: WB Saunders; 1992:43-60.
2. Shields JA. Tumors and related lesions of the retinal pigment epithelium. In: Guyer DR, Yannuzzi LA, Chang S, et al. Retina-Vitreous-Macula, Vol. 2. Philadelphia: WB Saunders, 1999;1116-1129.
3. Blair NP, Trempe CL. Hypertrophy of the retinal pigment epithelium associated with Gardner’s syndrome. Am J Ophthalmol 1980;90:661-667.
4. Shields JA, Shields CL, Shah PG, et al. Lack of association among typical congenital hypertrophy of the retinal pigment epithelium, adenomatous polyposis, and Gardner syndrome. Ophthalmology 1992;99:1709-1713.
5. Whitson WE, Orcutt JC, Walkinshaw MD. Orbital osteoma in Gardner’s syndrome. Am J Ophthalmol 1986;101;236-241.
6. Kunikata H, Abe T, Yoshida M, et al. The characteristics of congenital hypertrophy of retinal pigment epithelium in Turcot’s syndrome. Ophthalmologica 2000;214:374-375.
7. Aiello LP, Traboulsi EI. Pigmented fundus lesions in a preterm infant with familial adenomatous polyposis. Arch Ophthalmol 1993;111:302-303.
8. Kasner L, Traboulsi EI, Delacruz Z, et al. A histopathologic study of the pigmented fundus lesions in familial adenomatous polyposis. Retina 1992; 12:35-42.
9. Traboulsi EI, Murphy SF, de la Cruz ZC, et al. A clinicopathologic study of the eyes in familial adenomatous polyposis with extracolonic manifestations (Gardner’s syndrome). Am J Ophthalmol 1990;110:550-561.
10. Traboulsi EI, Maumenee IH, Krush AJ, et al. Congenital hypertrophy of the retinal pigment epithelium predicts colorectal polyposis in Gardner’s syndrome. Arch Ophthalmol 1990;108:525-526.
11. Traboulsi EI, Maumenee IH, Krush AJ, et al. Pigmented ocular fundus lesions in the inherited gastrointestinal polyposis syndromes and in hereditary nonpolyposis colorectal cancer. Ophthalmology 1988;95:964-969.
12. Krush AJ, Traboulsi EI, Offerhaus JA, et al. Hepatoblastoma, pigmented ocular fundus lesions and jaw lesions in Gardner syndrome. Am J Med Genet 1988;29:323-332.
13. Traboulsi EI, Krush AJ, Gardner EJ, et al. Prevalence and importance of pigmented ocular fundus lesions in Gardner’s syndrome. N Engl J Med 1987;316:661-667.
14. Heinemann MH, Baker RH, Miller HH, et al. Familial polyposis coli: the spectrum of ocular and other extracolonic manifestations. Graefes Arch Clin Exp Ophthalmol 1991;229:213-218.
15. Lewis RA, Crowder WE, Eierman LA, et al. The Gardner syndrome. Significance of ocular features. Ophthalmology 1984;91:916-925.
16. Munden PM, Sobol WM, Weingeist TA. Ocular findings in Turcot syndrome (glioma-polyposis). Ophthalmology 1991;98:111-114.
17. Ruhswurm I, Zehetmayer M, Dejaco C, et al. Ophthalmic and genetic screening in pedigrees with familial adenomatous polyposis. Am J Ophthalmol 1998;125:680-686.
18. Traboulsi EI, Apostolides J, Giardiello FM, et al. Pigmented ocular fundus lesions and APC mutations in familial adenomatous polyposis. Ophthalmic Genet 1996;17:167-174.
19. Rossato M, Rigotti M, Grazia M, et al. Congenital hypertrophy of the retinal pigment epithelium (CHRPE) and familial adenomatous polyposis (FAP). Acta Ophthalmol Scand 1996;74:338-342.
20. Aiello LP, Traboulsi EI. Pigmented fundus lesions in a preterm infant with familial adenomatous polyposis. Arch Ophthalmol 1993;111:302-303.
21. McKay DL. Congenital hypertrophy of the retinal pigment epithelium and familial adenomatous polyposis. Aust N Z J Ophthalmol 1993;21:3-6.
22. Romania A, Zakov ZN, McGannon E, et al. Congenital hypertrophy of the retinal pigment epithelium in familial adenomatous polyposis. Ophthalmology 1989;96:879-884.
23. Romania A, Zakov ZN, Church JM, et al. Retinal pigment epithelium lesions as a biomarker of disease in patients with familial adenomatous polyposis. A follow-up report. Ophthalmology 1992;99:911-913.
▪ Retinal Pigment Epithelial Hamartomas Associated with Familial Adenomatous Polyposis (RPEH-FAP)
Multifocal lesions of hypertrophy and hyperplasia of the RPE are known to be a hallmark of patients with familial adenomatous polyposis (FAP) and Gardner syndrome, familial conditions that predispose the patient to colorectal cancer. Gardner syndrome consists of FAP with extracolonic manifestations, like desmoid tumors, osteomas, and other benign tumors. Examples of RPEH-FAP are shown.
1. Romania A, Zakov N, McGannon E, et al. Congenital hypertrophy of the retinal pigment epithelium in familial adenomatous polyposis. Ophthalmology 1989;96:879-884.
2. Whitson WE, Orcutt JC, Walkinshaw MD. Orbital osteoma in Gardner’s syndrome. Am J Ophthalmol 1986; 101;236-241.
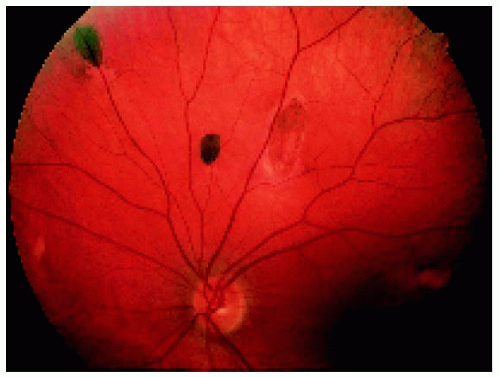 Figure 22.43. Multifocal fundus lesions. About six lesions are shown, some of which are pigmented and some nonpigmented. (Courtesy of Nicholas Zakov, MD.) |
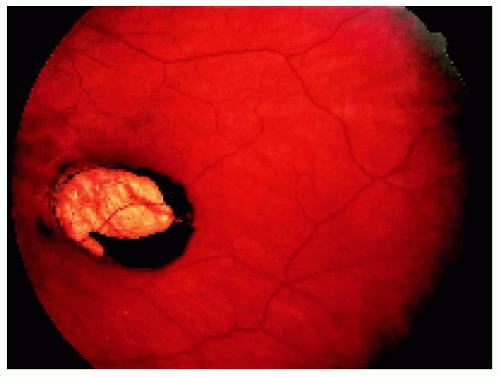 Figure 22.44. A larger lesion that is mostly depigmented. (Courtesy of Nicholas Zakov, MD.) |
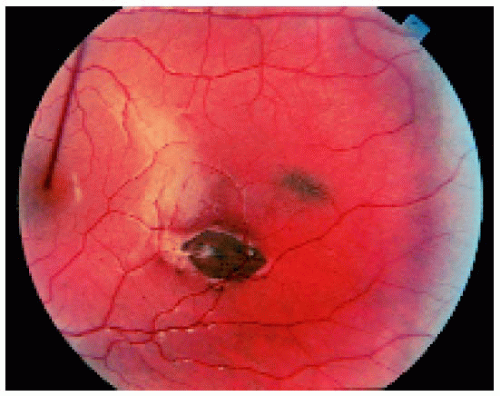 Figure 22.45. Two lesions in the macular region. |
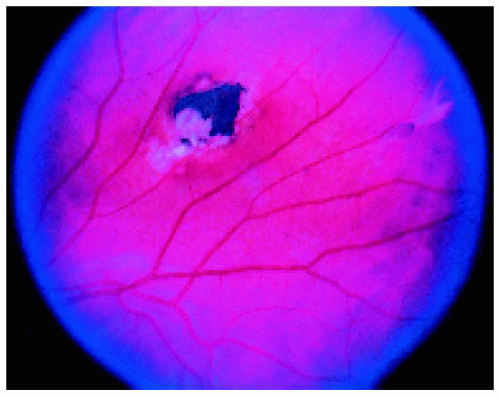 Figure 22.46. Typical fundus lesion in a patient with familial adenomatous polyposis. (Courtesy of Norman Blair, MD.) |
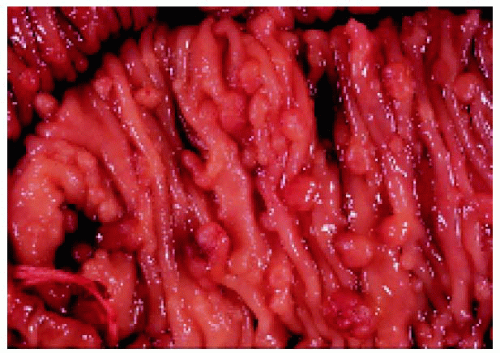 Figure 22.47. Sections of colon, showing numerous polyps in a patient who also had typical fundus lesions. (Courtesy of James Bolling, MD.) |
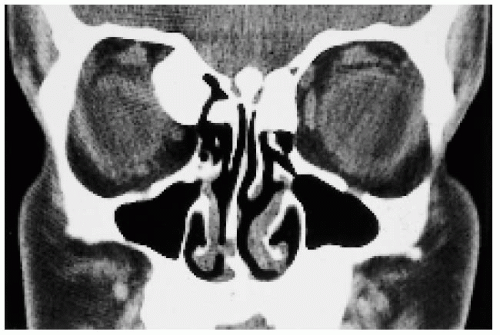 Figure 22.48. Coronal computed tomogram, showing orbital osteoma as part of Gardner syndrome. (Courtesy of James Orcutt, MD.) |
Pseudoneoplastic Reactive Hyperplasia of the Retinal Pigment Epithelium (RPE)
General Considerations
The RPE has a marked propensity to undergo reactive hyperplasia as a result of ocular insults such as inflammation or trauma. In most instances, RPE hyperplasia is small and typical and does not pose a diagnostic problem. On occasion, such RPE proliferation can simulate a pigmented neoplasm such as choroidal or ciliary body melanoma or true neoplasms of the RPE (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13).
Clinical Features
Pseudoneoplastic reactive hyperplasia of the RPE can assume a number of clinical appearances. It can appear as a diffuse or sessile lesion or as a nodular mass. Its dark black color and associated signs of prior inflammation or trauma can help to differentiate the lesion from choroidal melanoma. Secondary retinal detachment is uncommon. Even though this lesion is relatively stable, a few cases have been observed in which small focal areas of RPE hyperplasia have shown evolution into a nodular tumor that transgresses the retina into the vitreous cavity (12). In rare instances, presumed reactive hyperplasia of the RPE can even extend extrasclerally (13).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


