Fig. 8.1
This pedigree represents an apparently sporadic bilateral retinoblastoma in the proband, but genetic testing of the parent revealed that the unaffected mother is mosaic for the RB1 mutation. In this case, the mother must be counseled regarding her own reproductive and cancer risks. This case highlights the importance of testing all unaffected parents for the germline mutation found in their child
8.6 Confounding Factors
Many confounding factors can complicate what may seem to be a simple pedigree or an apparently nonfamilial case. When these scenarios are understood, the counselor will be better prepared to avoid these common pitfalls.
8.6.1 Chromosome 13q14 Deletions
Deletion of chromosome 13q14, the locus of the RB1 gene, and neighboring regions on the long arm of chromosome 13 can lead to intellectual disability and retinoblastoma. The size of the deletions varies and the phenotype is also variable. However, the correlation between the size of the deletion and the severity of the clinical phenotype has not been established. Some individuals with small deletions of this area are developmentally normal. In children with a deletion involving chromosome 13q14, developmental delay or intellectual disability may be appreciated before retinoblastoma is diagnosed (Fig. 8.2).
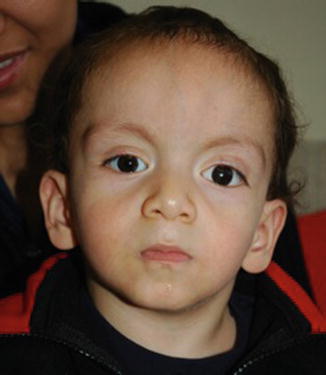
Fig. 8.2
This boy was referred to a pediatric neurologist for hypotonia before his retinoblastoma was diagnosed. He has a chromosome 13q deletion visible on routine banding. He has an ocular prosthesis following enucleation of his left eye for retinoblastoma. He also has developmental delay and mild facial dysmorphism: broad forehead with frontal bossing, arched eyebrows, hypertelorism, small mouth
Children with chromosome13q14 deletions may develop retinoblastoma at a somewhat later age and often they develop only one tumor. One may speculate that when the “first hit” is a large deletion, gene conversion, a common pathway for the “second hit” in retinoblastoma, may lead to premature cell death instead of cancer. Paradoxically by reducing cell viability with the “second hit,” large mutations can act as low-penetrance mutations.
In our center, all children have high-resolution chromosome analysis and fluorescence in situ hybridization (FISH) for 13q14 as part of their initial evaluation to rule out both macroscopic and microscopic deletions [3]. When a chromosome deletion is discovered in an affected patient, a microarray should be done to determine the size and extent of the deletion. The associated intellectual disability has been mapped to the nearby NUFIP1 and PCDH8 genes. The parents should also have testing with microarray or FISH for 13q14 to rule out a similar deletion, inversion, or other heritable chromosome anomaly.
8.6.2 Mosaicism
Mosaicism, in which the RB1 mutation is present in some but not all cells of the affected child, is common in the first affected member of a family with retinoblastoma [4–10]. Mosaics for RB1 germline mutations often have unilateral retinoblastoma and later onset and they lack a family history of the disease. However, mosaicism has also been seen in patients with bilateral retinoblastoma and in unaffected parents of affected children. There is no technique that will reliably detect all cases of mosaicism. Gene sequencing may miss mosaicism when less than 20 % of cells have a mutation. Even when more than one tissue is studied, mosaicism can never be completely ruled out in the first affected member of the family. When a child with unilateral sporadic retinoblastoma has normal RB1 gene test results, the chance of a germline mutation is never zero. There is always a small residual risk for undetected mosaicism. The counseling session should include a discussion of the possibility of low-level germline mosaicism.
A genetic counselor can anticipate mosaicism in a multi-generation retinoblastoma family when the first affected individual, the “founder,” has unilateral retinoblastoma or a late-onset tumor yet their affected offspring have bilateral, early-onset retinoblastoma. In a retinoblastoma family with 2 or more affected generations, including a parent and child, it is always best to start the testing process in the child from the second affected generation. This avoids the possibility of a false-negative result due to undetected mosaicism in the first affected member of the family. Mosaicism, when it occurs, is limited to the first affected member of the pedigree. Mosaicism is not hereditary. The affected child of a mosaic individual inherits the deleterious mutation and is not mosaic. Such a family is illustrated in Fig. 8.3. The mother had unilateral retinoblastoma and normal RB1 gene analysis, while her daughter, with bilateral retinoblastoma, had a detectable RB1 mutation. Later the same mutation was demonstrated in the mother’s blood using a specific technique for that mutation: PCR with an allele-specific oligonucleotide probe. This family also shows the value of testing more than one individual in a family when a germline mutation is expected.

Fig. 8.3
The mother in this photograph had unilateral retinoblastoma. After her daughter was diagnosed with bilateral retinoblastoma, the mother was found to be mosaic for the germline mutation in RB1 that caused the daughter’s disease. This family illustrates the point that when retinoblastoma is caused by a new sporadic mutation, the founder individual may be mosaic for the RB1 mutation (i.e., it can occur at some point after conception and the first cell division). When searching for the mutation in a two-generation family, the second generation should always be tested first, if possible. Also this family confirms comments in the text that a negative DNA test for a germline mutation in a unilateral patients may fail to detect low-level mosaicism
8.6.3 Low-Penetrance Mutations and Variable Expressivity
Low-penetrance mutations, often due to missense mutations in RB1 that do not truncate the protein product, and variable expressivity are well documented in the retinoblastoma literature [11–13]. The specific type of RB1 mutation is important in determining whether to expect complete penetrance, high penetrance, or variable penetrance. Penetrance varies with the in-frame (low penetrance) or out-of-frame (high penetrance) nature of splice site mutations. Another mechanism for variable penetrance occurs when exon 1 mutations produce functional transcripts through alternate mRNA transcription [14, 15]. We stress the need for parental dilated eye exams before genetic counseling so a retinocytoma/retinoma (Chap. 7) that may be present but previously unknown is documented prior to counseling. A parent who is unaffected and healthy can share the same RB1 germline mutation with their affected child. So-called pseudo low penetrance may also lead the counselor astray. This refers to two affected relatives in a large pedigree giving the appearance of familial retinoblastoma, when in fact, the retinoblastoma tumors arose from independent and unrelated RB1 mutations [16].
8.6.4 Family History of Other Early-Onset Cancers
The family history is often positive for early-onset cancers of different types. Otherwise healthy parents and grandparents of patients with isolated retinoblastoma may have rare or multiple cancers suggesting the possibility of other cancer predisposing genetic disorders in the family. This can complicate genetic counseling for the family of a child with sporadic unilateral retinoblastoma. In the face of normal RB1 mutation analysis, the genetic counselor might modify the risk for subsequent cancers based on the family history of other cancers. We have observed early-onset melanoma in the unaffected daughter of a mother with retinoblastoma who had died of gastric cancer. Conversely, we have also observed early-onset melanoma in the otherwise normal mother of a boy with isolated sporadic retinoblastoma whose RB1 mutation analysis was normal. In both of these families, cancer surveillance and high-risk follow-up were recommended.
8.6.5 Evolving Phenotypes and Changing Pedigrees
Evolving phenotypes and changing pedigrees sometimes make it necessary to revise risks and re-counsel families. After counseling a patient with unilateral sporadic retinoblastoma, the counselor may have to revise their risk assessment after the discovery of a second affected individual in the family or a second tumor in the proband. This is a special concern when counseling the parents of a young infant with a unilateral tumor.
Counseling should always include the parents, whose status may change as a result of genetic testing. An unaffected parent may be surprised to find that they harbor a germline mutation and are at increased risk for non-ocular cancers. This parent now needs to be monitored and counseled. It is also important to counsel the parent who had unilateral Rb themselves and but did not know their RB1 mutation status until the birth of a child with bilateral Rb. This affected parent is now clearly a germline mutation carrier and at risk for secondary tumors. In one such family, the father had unilateral Rb and his child had bilateral Rb. The father was unaware of his own risk for secondary tumors. After genetic counseling, he was examined at our recommendation and was diagnosed with a melanoma even while his child was still undergoing treatment. This illustrates the need to consider the affected parent as a patient, even at a time when most of the medical attention is focused on the affected child.
8.6.6 Intellectual Disability
All children with retinoblastoma should be monitored for age-appropriate developmental milestones. However, intellectual disability in children with retinoblastoma is not always due to a deletion of chromosome 13q14. Those who are developmentally delayed deserve a prompt and thorough evaluation. The correct diagnosis can be delayed when the team attributes developmental delay to a 13q deletion but fails to obtain chromosome analysis and fluorescence in situ hybridization studies or microarray studies. We have seen a child with retinoblastoma and autistic features, who had Fragile X syndrome. Other patients have intellectual disability of unknown cause without a clear causal link to their retinoblastoma.
8.6.7 Congenital Anomalies
Congenital anomalies are commonly encountered in patients with retinoblastoma. In our clinic, we have seen retinoblastoma patients with clubfeet, dextrocardia, ear anomalies, etc. It is unclear whether there are more than the expected number of congenital anomalies among children with retinoblastoma because there have been no studies. However, in our center, more than the expected 3 % of affected children have congenital anomalies. Although this could be due to ascertainment bias, it also raises questions about common environmental or genetic/epigenetic causation for retinoblastoma and birth defects. In either case, the presence of other anomalies further complicates the genetic counseling process. A complete genetic assessment is important for any child with retinoblastoma and unexpected findings or developmental delay.
8.7 The Isolated Case of Unilateral Retinoblastoma
It is the isolated case of unilateral retinoblastoma that is most problematic for the genetic counselor. The lack of a family history and an older age at onset of unilateral sporadic retinoblastoma do not exclude a germline RB1 mutation [17]. We recently found a germline RB1 mutation in a child with unilateral sporadic retinoblastoma who was diagnosed at age 5 years. Although most children with unilateral retinoblastoma will not have heritable retinoblastoma, a significant minority will have a germline mutation in the RB1 gene. These are the children and families who may benefit the most from genetic counseling since some of these children receive more intervention than they need and others receive too little. For now, when a germline RB1 mutation is diagnosed, we can only modify surveillance and advice about reproductive risks. Reducing the morbidity and mortality associated with retinoblastoma is an important goal of genetic counseling. Eventually, we hope to have effective strategies to reduce cancer risk in individuals who carry RB1 germline mutations. We hope that all children with unilateral sporadic retinoblastoma will be able to undergo RB1 testing to determine their germline mutation status.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


