Chapter 42 Retinoblastoma
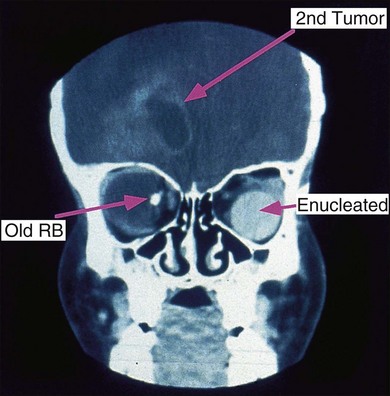
Retinoblastoma is an uncommon malignant ocular tumor of childhood, occurring in 1 : 18 000 live births.1 Late diagnosis globally results in up to 70% mortality; where optimal therapy is accessible, more than 95% of children are cured. An integrated team approach of clinical specialists (ophthalmologists, pediatric oncologists and radiotherapists, nurses, geneticists) with imaging specialists, child life (play) specialists, parents, and others is an effective way to manage retinoblastoma. National guidelines can bring the whole health team up to developed standards and set the stage for audits, studies, and clinical trials to continuously evolve better care and outcomes.2 The tumor(s) arises from embryonic retinal cells so the majority of cases occur under the age of 4 years. Primary treatments include enucleation and chemotherapy with laser and cryotherapy. Patients with a constitutional mutation of the RB1 tumor suppressor gene are at increased life-long risk of developing other cancers, which is increased with exposure to radiation (Figs 42.1 and 42.2).3,4 Therefore, radiation is no longer a primary therapy to save an eye, and screening for extraocular and trilateral retinoblastoma is performed with MRI and ultrasound, not CT scan.
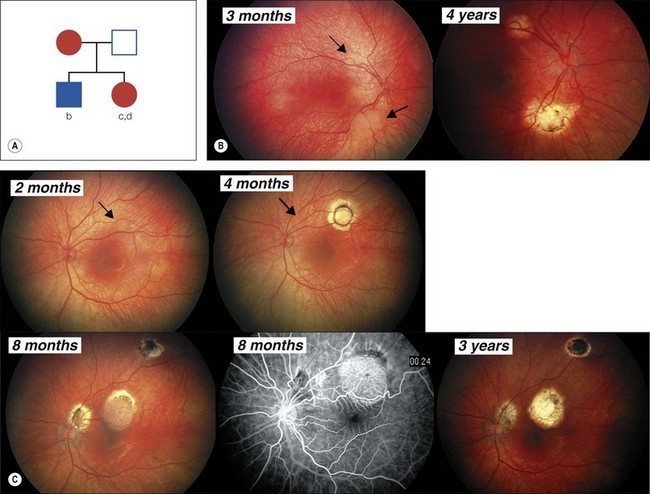
The study of retinoblastoma has been seminal in the understanding of cancer in general. Studies of retinoblastoma have revealed that hereditary and non-hereditary tumors are initiated by the loss of both alleles of the tumor suppressor gene, RB1.5,6 The existence of specific genes that act to suppress cancer was predicted from clinical studies of retinoblastoma.7,8 The RB1 gene was the first tumor suppressor gene to be cloned,5 and has been found to have a critical role in many types of cancer.
Pathogenesis of retinoblastoma
Heritable and non-heritable retinoblastoma
All children with retinoblastoma tumors in both eyes (bilateral) have an RB1 gene mutation on one of their chromosomes (13) that predisposes them to develop retinal tumors in infancy and other cancers throughout life (see Figs 42.1 and 42.2). While 90% have no family history of retinoblastoma and are the first affected in their family with a new germ line mutation,9 50% of their offspring will inherit the mutant RB1 gene and develop tumors. Most children without a family history with retinoblastoma in only one eye have normal constitutional RB1 alleles, but the eye tumor(s) loses both functional alleles, similar to hereditary tumors. Fifteen percent of persons who had unilateral retinoblastoma have constitutional RB1 mutations that can be transmitted to their offspring. Molecular and clinical genetics is an integral part of the management of all families affected by retinoblastoma.
Loss of both RB1 alleles induces retinoblastoma
The observation that the children with bilateral retinoblastoma tend to be diagnosed at a younger age than those with non-hereditary retinoblastoma led to Knudson’s prediction that two mutational events were required to initiate retinoblastoma tumors.7 His analysis suggested that in the presence of a predisposing constitutional mutation a single second mutation in one developing retinal cell initiated tumor development (heritable retinoblastoma), but both alleles were mutated in the single developing retinal cell in non-heritable unilateral retinoblastoma. The two events could be mutations of both alleles of a gene that would “suppress” tumor formation in the retina.8 The chance of losing the second RB1 allele from developing retinal cells with only one normal RB1 allele is sufficiently high that multiple tumors are common in hereditary retinoblastoma (see Fig. 42.1). However, it is virtually impossible for children without constitutional RB1 mutations to lose both alleles from several retinal cells so they develop only one, unilateral tumor (Fig. 42.3), and tend to be diagnosed at an older age than children with hereditary retinoblastoma.
Function of the retinoblastoma protein
The product of the RB1 gene (pRB) is a 110 kDa phosphoprotein that interacts with many proteins in the regulation of the cell cycle, differentiation, and control of genomic stability.10 DNA tumor viruses that induce cancer, such as human papilloma virus, do so in part by binding to pRB through the “pocket” region of pRB.
Germ line mutation of RB1 leads to a 40 000-fold relative risk (RR) for retinoblastoma, a 500-fold RR for sarcoma that is increased up to 2000-fold by therapeutic radiation, but no increase in the RR for leukemia.11 Although pRB is key to all cycling cells, its function in development is highly tissue-specific. A subset of developing retinal cells may be uniquely dependent on pRB in order to differentiate terminally into adult, functioning retina. Loss of pRB promotes genomic changes and instability, leading to further mutations in oncogenes and other tumor suppressor genes that result in a retinal tumor.12,13
Spectrum of RB1 mutations
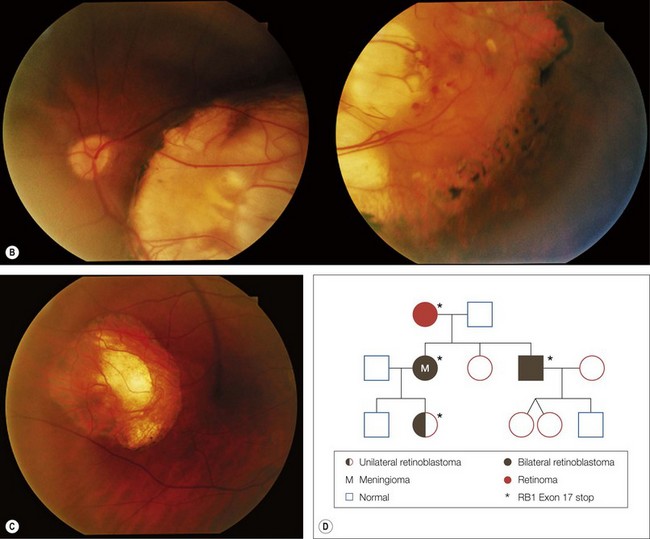
The majority of RB1 mutations are unique to each family, and are distributed throughout the RB1 gene with no real hot spots.9 Sensitive mutation identification requires determination of the copy number of each exon and the gene promoter to reveal large deletions and duplications, sequencing for point mutations, examination of the mRNA to confirm or detect intronic mutations altering exon splicing, and assay for the methylation status of the promoter in tumor samples (Fig. 42.4). Application of these techniques, combined with a retinoblastoma-specific focused expertise in interpreting the data, identifies over 95% of the RB1 mutations9,14 (see Figs 42.1–42.4).
Other manifestations of RB1 mutant alleles
Mutation of RB1 also predisposes to benign retinal tumors, retinoma,15 ectopic intracranial retinoblastoma (trilateral retinoblastoma),16,17 and second non-ocular malignancies.18,19
Retinoma
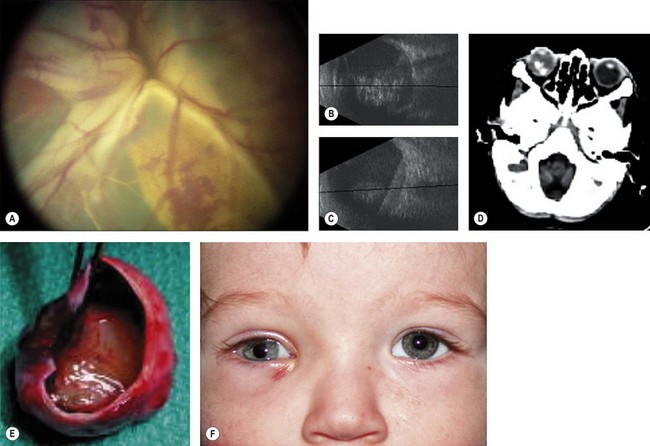
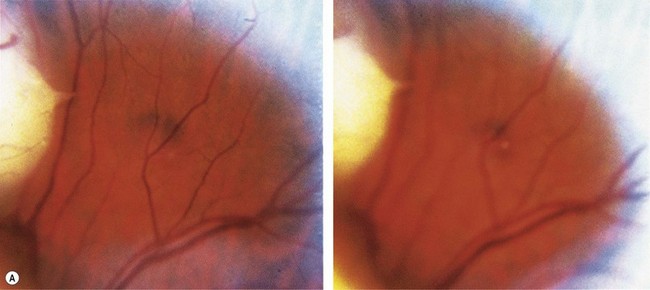
A retinoma is a non-malignant manifestation of the RB1 mutation.15 Three features characterize these non-progressive lesions: an elevated grey retinal mass, calcification, and surrounding retinal pigment epithelium (RPE) proliferation and pigmentation (Fig. 42.5). These features are also seen after radiation treatment for retinoblastoma. If documented in childhood, which is very rare, retinoma is a quiescent tumor that has not progressed to malignancy. Occasional cases occur where a retinoma progresses to active retinoblastoma. However, retinoma commonly underlies active retinoblastoma and can be discovered on pathologic examination of an enucleated eye.12 A distinctive feature is fleurette formation and absence of proliferative markers. Both RB1 alleles are mutant in the retinoma and genomic instability is detectable, which progresses in degree and number of genes involved in the adjacent highly proliferative retinoblastoma.12 Discovery of retinoma on retinal examination of a relative of a patient with retinoblastoma indicates that they carry the RB1 mutant allele (see Fig. 42.5).
Ectopic intracranial (trilateral) retinoblastoma
Trilateral retinoblastoma is a midline intracranial tumor or a primary pineal tumor associated with heritable retinoblastoma that is not related to a metastasis.16 The tumors are neuroblastic and resemble a poorly differentiated retinoblastoma. Pineal tumors arise in 5% of children with an RB1 mutation but should not be confused with pineal cysts which occur in 2% of all children and require no treatment.20 Affected children may present with raised intracranial pressure and are found to have a pineal or parasellar mass on MRI.17 Routine screening by MRI for intracranial tumors may detect pineal tumors at a stage when they can be cured.16,17
Multiple different malignancies
Persons with RB1 gene mutant alleles are at increased risk of developing second non-ocular malignancies4,18,19 which may occur within or outside the radiation field (see Fig. 42.2). Radiation, particularly of infants under 1 year of age, increases the risk of sarcomas and other cancers within the radiation field. Osteosarcoma is the commonest second primary tumor in persons with RB1 mutations, but a wide variety of other neoplasms have been reported. Since these radiation-induced tumors are very difficult to treat, in the past more children with RB1 mutations have died of their second tumor than have died of uncontrolled retinoblastoma. Radiation is now restricted to salvage of the remaining eye in children with retinoblastoma.21
Genetic counseling for retinoblastoma
The most accurate way to predict who in a family will develop retinoblastoma is to test them for the precise RB1 mutant allele found in the proband. In the absence of precise knowledge of the RB1 mutant alleles in tumor or blood, the empiric risk for the relatives of retinoblastoma patients to be affected can be estimated.22 Offspring of patients with a family history of retinoblastoma or bilateral tumors have a 50% risk of inheriting the mutant allele and a 45% risk of developing retinoblastoma, due to incomplete penetrance. When two affected children are born to apparently normal parents, one parent must be carrying but not expressing the mutant allele. Hence, there is also a 45% risk that any subsequent child born will develop retinoblastoma. The risk that other relatives have inherited the mutant allele depends on the number of intervening “apparently normal” individuals, each of which have a 10% chance of carrying but not expressing the mutant allele. The risk falls by a factor of 0.1 for each intervening unaffected generation. Since 15% of patients with unilateral retinoblastoma have a germinal mutation, the offspring of individuals with unilateral retinoblastoma have a 7.5% risk of carrying the abnormal gene. The probability of other relatives developing retinoblastoma falls by a factor of 0.1 for each intervening unaffected generation.22
Infants born with a risk of developing retinoblastoma need to be examined immediately after birth and then at regular intervals to detect early tumors that can be treated to obtain the best visual result (see Fig. 42.1). Infants proven to carry the family’s RB1 mutant allele can be delivered a few weeks early, to optimize the chance to keep good vision with minimally invasive therapy. Examination of the retina starts at birth, and continues at frequent intervals depending on the child’s risk. Up to 3 months of age, examination may be done without general anesthetic, greatly facilitated by the RetCam® camera on video mode. After 3 months, anesthetic is necessary to get an accurate view of the retina to detect tiny tumors up to the ora serrata.
Timely and sensitive molecular diagnosis of RB1 mutations has a strong positive effect on quality of outcomes: early treatment of retinoblastoma achieves lower risks and better health outcomes, allows families to make informed family-planning decisions, and costs less than conventional surveillance.9,23 The savings when at risk children avoid repeated examinations substantially exceeds the one-time cost of molecular testing. Moreover, health care savings continue to accrue as succeeding generations avoid the unnecessary examinations and often do not need molecular analysis because their parents do not carry the family’s mutant allele.
The RB1 mutations usually result in unstable or absent protein. Such mutations show high penetrance (> 95% of offspring affected) and expressivity (average of seven tumors per child). More uncommon RB1 mutations cause lower penetrance and expressivity:23 “In frame” deletions or insertions that result in a stable but defective pRB;24 promoter mutations that result in a reduced amount of otherwise normal protein;23 and splice mutations that may be additionally altered by unlinked “modifier genes”.25
Presentation
The majority of children with retinoblastoma without a family history are first noticed because of leukocoria (Table 42.1).26 Parents observe an odd appearance in their child’s eye. Too often primary health care personnel are unaware of the importance of what the parents are saying and diagnosis is delayed. Health care professionals should respond to a parent’s description of a “cat’s eye reflex” by referring the child for full investigation of the eyes (Fig. 42.7).
Table 42.1 Presenting symptoms and signs of retinoblastoma (Ellsworth 1969)
| White reflex | 56% |
| Strabismus | 20% |
| Glaucoma | 7% |
| Poor vision | 5% |
| Routine examination | 3% |
| Orbital cellulitis | 3% |
| Unilateral mydriasis | 2% |
| Heterochromia iris | 1% |
| Hyphema | 1% |
| Other | 2% |
Ellsworth RM. The practical management of retinoblastoma. Trans Am Ophthalmol Soc 1969; 67: 462–534.
Retinoblastoma family support groups have embarked upon awareness campaigns to educate the lay public in the importance of a “white pupil” (Figs 42.6 and 42.7). Digital images of the baby with retinoblastoma frequently show a white pupil, “photoleukocoria,” in contrast to the red eye reflex of the flash picture of a normal eye.27 While retinoblastoma is the most important and dangerous condition to cause leukocoria, various conditions also show unusual appearance on flash images, such as congenital cataract, myelinated nerve fibers, optic nerve coloboma, high myopia, astigmatism, and normal optic nerves when the camera angle is directed at the optic nerve.28
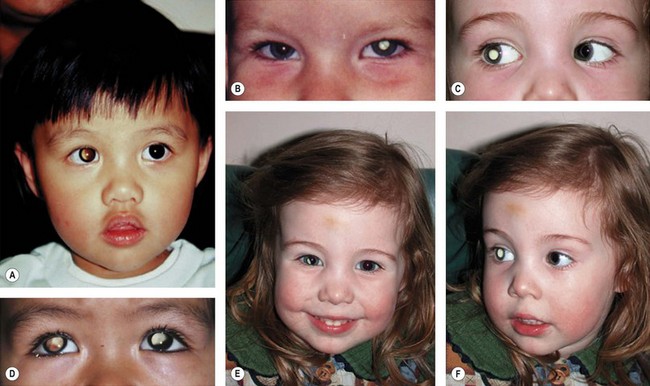

Fig. 42.7 Childhood Eye Cancer Trust awareness campaign poster (UK).
(This image is part of a campaign run by the Childhood Eye Cancer Trust UK.)
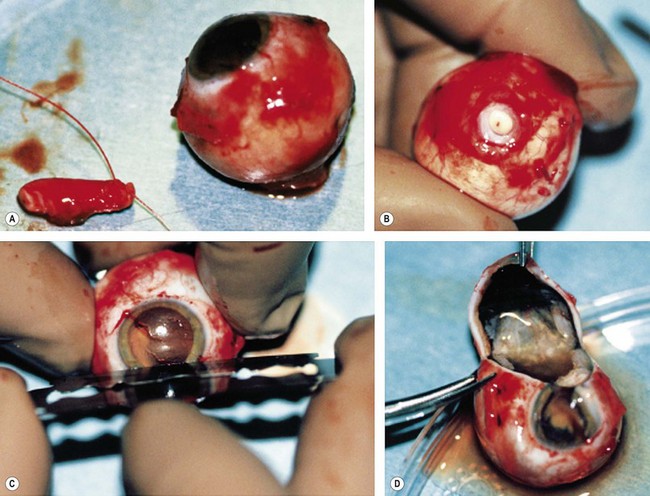
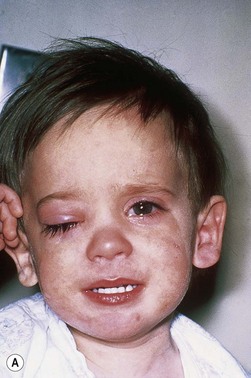
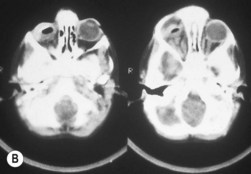
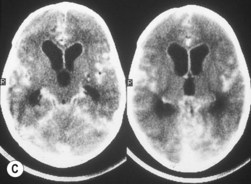
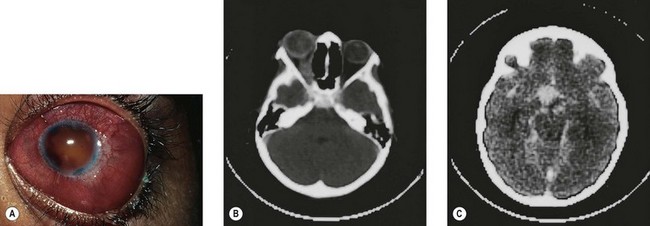
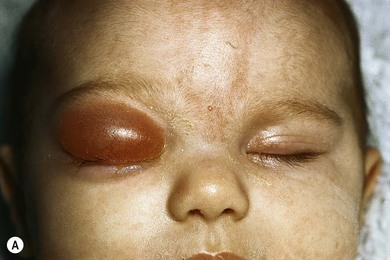
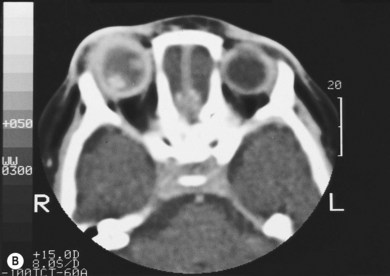
The second most common presenting sign of retinoblastoma is strabismus (esotropia or exotropia).26 The red reflex test should be applied to any child with strabismus or suspected strabismus, with prompt, urgent referral from the primary health level to an ophthalmologist if the red reflex test is abnormal.2 Other presenting symptoms and signs (see Table 42.1) include a painful red eye (due to glaucoma), orbital cellulitis secondary to extensive necrosis of the intraocular tumor (Figs 42.8–42.10),29 unilateral mydriasis, heterochromia, hyphema, hypopyon, uveitis, and “searching” nystagmus (due to blindness from bilateral macular involvement).26 In countries with limited medical services, many children present with extensive unilateral or bilateral proptosis with orbital extension and/or metastatic disease due to delayed access to care (see Figs 42.8 and 42.9).
Retinoblastoma in babies and children that are relatives of patients with heritable retinoblastoma should be looked for specifically by screening examinations, before any symptoms occur unless genetic testing rules out the mutant allele in that individual (see Fig. 42.1). For most families, it is possible to detect the precise RB1 mutation of the proband, check the relatives for that mutation, identify those carrying the mutant allele, and diagnose and initiate treatment early when the tumors are small and can often be cured by laser therapy alone, or with short cycles of chemotherapy in order to obtain the best visual outcome.
Diagnosis
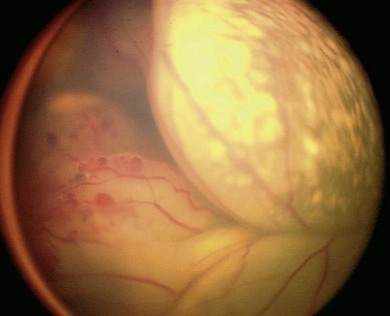
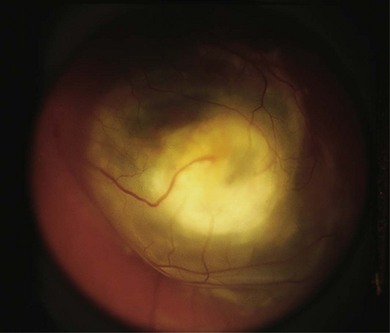
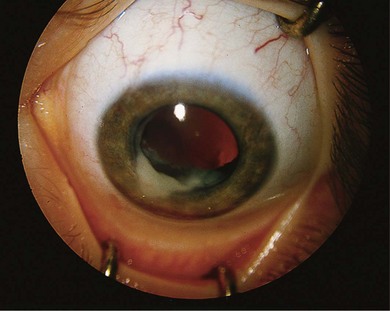
The initial examination of the child presenting as possible retinoblastoma will provide a short-list of differential diagnoses, including Coats’ disease (Fig. 42.11), persistent hyperplastic primary vitreous, toxocara (Fig. 42.12), medulloepithelioma (Fig. 42.13), and others (Box 42.1).26 Referral of a child with possible retinoblastoma is urgent, generally requiring examination within 1 week.2
Box 42.1
Modified from Shields JA, Augsburger JJ. Current approaches to diagnosis and management of retinoblastoma. Surv Ophthalmol 1981; 25: 347–72.
Differential diagnosis of retinoblastoma
| Hereditary conditions | Inflammatory conditions |
| Norrie’s disease | Toxocariasis |
| Warburg’s syndrome | Toxoplasmosis |
| Autosomal recessive retinal dysplasia | Metastatic endophthalmitis Viral retinitis Vitritis Tumors Astrocytic hamartoma Medulloepithelioma Choroidal hemangioma Combined hamartoma |
| Dominant exudative vitreoretinopathy | |
| Juvenile X-linked retinoschisis | |
| Orbital cellulitis | |
| Developmental anomalies | |
| Persistent hyperplastic primary vitreous | |
| Cataract | |
| Coloboma | |
| Congenital retinal fold | |
| Myelinated nerve fibers | |
| High myopia | |
| Morning glory syndrome | |
| Others | |
| Coats’ disease | |
| Retinopathy of prematurity | |
| Rhegmatogenous retinal detachment | |
| Vitreous hemorrhage | |
| Leukemic infiltration of the iris |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


