Fig. 9.1
Histological section of the retina, the retinal pigment epithelium, choroid, and sclera
The Müller cell endings connect photoreceptors to each other but also interlock with the microvilli of the retinal pigment epithelium. The outer segments of the photoreceptor cells (rods and cones) are embedded into the RPE microvilli depending on the light adaptation. Photoreceptor outer segment renewal takes place by the shedding of the tips with subsequent phagocytosis by RPE cells where lysosomal degradation and, for some molecules, recycling to photoreceptors take place.
The outer nuclear layer consists of the nuclei of the rods and cones and is located just external to the outer plexiform layer. The nuclei of the cones are located closer to the external limiting membrane than the rod nuclei are. The outer two thirds of the outer plexiform layer consist of axons of the photoreceptor cells, while the inner third contains the synaptic connections between the photoreceptor axons and the axons of the bipolar, horizontal, and amacrine cells that comprise the inner nuclear layer. This cellular/synaptic complex is located approximately in the center of the retina and is an important physical barrier for exudates and defines the outermost extent of the deep retinal capillary plexus that lies approximately parallel to the vascular network in the nerve fiber layer.
Internal to the outer plexiform layer is inner nuclear layer, which consists of the cell bodies of bipolar, amacrine, horizontal, and Müller cells. This layer is richly perfused by retinal capillaries. The nuclei of the bipolar and Müller cells are located in the middle layers of the inner nuclear layer, whereas the nuclei of horizontal cells lie towards the outer plexiform layer and the amacrine cells towards the inner plexiform layer. The inner plexiform layer is composed of the axons of the bipolar and amacrine cells, the dendrites of the ganglion cells, and their interconnecting synapses. The ganglion cell layer is composed of multiple cell layers only in the macular area, whereas is composed of a cellular monolayer outside the macular area. The nerve fiber layer is composed of nonmyelinated axons of the ganglion cells. The inner limiting membrane (ILM) is a basement membrane in which the footplates of Müller cells are embedded. Its inner face is smooth, and the outer aspect is irregular and varies in thickness across the retina. It is thinnest near the optic disk and overlying the retinal vessels.
9.2.1.1 The Macula
The macula has a diameter of approximately 5 mm and is located within the temporal vascular arcades. It includes the perifovea, the parafovea, the fovea, the foveola, and the central umbo. Histologically, the macula differs from the peripheral retina by having a multilayered arrangement of ganglion cells, which is thickest at the fovea where there is the higher density of cones. The foveola forms the center of the fovea and macula and has a diameter of 0.35 mm. Due to the lateral displacement of the inner retina and the fact that the umbo and the foveola contain only cones and Müller cells, its thickness is only 130 μm.
The axons of the cones at the fovea are called Henle fibers and are displaced laterally in an oblique fashion so that they border directly to the vitreous cavity. This is associated with a lateral displacement of the second and third neurons. Macular pigment binds particularly strongly within the Henle fiber layer.
9.2.1.2 The Retinal Pigment Epithelium (RPE)
Immediately underneath the neurosensory retina, in close contact with the photoreceptor outer segments, lies a monolayer of approximately one million polarized pigmented retinal pigment epithelial cells (RPE). In contrast to the sensory retina, this layer has no photoreceptive neural functions but is essential for the function and metabolic health of the photoreceptors. The RPE cells contain numerous melanin granules in the apical cytoplasm. They are cuboidal and hexagonal viewed en face and vary in size from 10 μm diameter at the macula to 60 μm in diameter in the periphery, where they are also flatter. RPE cells are largely postmitotic cells that decline in density throughout life, especially at the periphery. In humans there is no apparent RPE renewal by mitosis, although inflammation or trauma can cause a temporary reactive mitotic response. The RPE only extremely rarely becomes neoplastic, but often in pathological processes, it becomes metaplastic, forming, for instance, myoepithelial cells, fibrous scar tissue, or even bone. The apical region of the RPE cells is directed towards the photoreceptors, whereas the basal portion is attached to Bruch’s membrane and separated from it only by its thin basement membrane. The cell nucleus and mitochondria are located in the base of the cell. At the retinal periphery close to the ora serrata, RPE cells often can be multinucleated. It has been proposed that in that region RPE stem cells are present. Abundant mitochondria within the RPE reflect the high metabolic capacity of the cell. RPE cells are bound together by tight junctions, which are located near the cell apex (Verhoeff’s membrane). These junctions form the outer retinal blood–retinal barrier, whereas the inner retinal blood–retinal barrier is formed by the endothelium of the intraretinal blood vessels.
9.2.1.3 Bruch’s Membrane
Bruch’s membrane lies between the RPE and the choroid. Passing progressively towards the choroid from the RPE, Bruch’s membrane is composed of the RPE basement membrane, an inner collagenous layer, an elastic layer, an outer collagenous layer, and the basement membrane of the endothelial cells of the choriocapillaris.
9.3 Congenital Anomalies
Definition
Congenital anomalies of the retina fall into two general categories, stable structural or developmental anomalies and progressive inherited retinal degenerations. These latter conditions are usually the result of single or multiple genetic mutations. A list of stable structural or developmental anomalies is shown in Table 9.1. By definition, the structural and developmental lesions are not progressive disease processes. However, secondary changes can occur due to degeneration associated with the congenital condition, e.g., retinal detachment due to persistent fetal vasculature or choroidal neovascularization associated with macular telangiectasia.
Table 9.1
Structural developmental anomalies
Vascular |
Cavernous hemangioma of the retina |
Macular telangiectasia |
Coats’ disease |
Neuronal |
Albinism |
Color blindness |
Combined hamartoma |
Congenital hypertrophy of the RPE |
Retinal dysplasia |
Vitreous |
Persistent fetal vasculature |
Cyst |
The inherited conditions are for the most part progressive disorders. The age at which a disease phenotype appears is variable. Despite the hundreds of individual gene defects, there are certain stereotypical tissue responses that characterize progressive retinal degeneration: (1) photoreceptor atrophy associated with RPE hyperplasia, (2) photoreceptor atrophy combined with RPE atrophy, and (3) production of abnormal extracellular matrix. A sampling of inherited retinal disorders is listed on the basis of these tissue responses and is provided in Table 9.2. These tissue responses will be illustrated in the histopathology section below.
Table 9.2
Inherited retinal degenerations
Retinal atrophy and RPE hyperplasia |
Retinitis pigmentosa |
Neuronal ceroid lipofuscinosis |
Stargardt disease (severe phenotype) |
Retinal atrophy and RPE atrophy |
Gyrate atrophy |
Choroideremia |
Cone dystrophy |
Stargardt disease (moderate phenotype) |
Abnormal extracellular matrix |
Sorsby’s fundus dystrophy |
Best disease |
Stargardt disease (mild phenotype) |
Epidemiology
Structural or developmental anomalies of the retina are rare. Of these, abnormalities of the vasculature are by far the most common. The prevalence of persistence of the fetal vasculature is quite variable depending on how it is defined. Very mild persistence occurs in almost all infants with the last vestiges of the tunica vasculosa lentis regressing in the first few days after birth. However, more severe cases of persistent fetal vasculature are quite rare.
Inherited retinal degenerations are also uncommon. The prevalence of typical retinitis pigmentosa is approximately 1:5,000 worldwide. The incidence of Stargardt disease is 1:8,000 to 1:10,000. Retinitis pigmentosa and Stargardt disease are the most common of the progressive inherited retinal degenerations.
Clinical Features
The clinical features of the structural or developmental anomalies vary considerably based on what type and location of tissue are involved with the anomaly. As mentioned above, the most common of the congenital structural anomalies is persistent fetal vasculature [5]. During fetal development, there is a system of vessels that extends through the vitreous cavity and surrounds the lens. This vasculature system is called the hyaloid vasculature. With normal retinal development, these vessels regress. However, in some instances, there is persistence of these vessels. The clinical findings are quite variable based on the extent of the remaining vessels. In some instances, only a small dot-like cataract is present on the posterior surface of the lens, a so-called Mittendorf dot. However, in some cases there is persistence of the fetal vasculature on the optic nerve, and these persistent vessels can exert traction on the peripapillary retina (Fig. 9.2). Persistence of the fetal vasculature is also associated with cataract. In very severe cases, eyes with persistent fetal vasculature may be enucleated due to poor vision and concern that the changes in the eye are due to an intraocular malignancy (retinoblastoma).
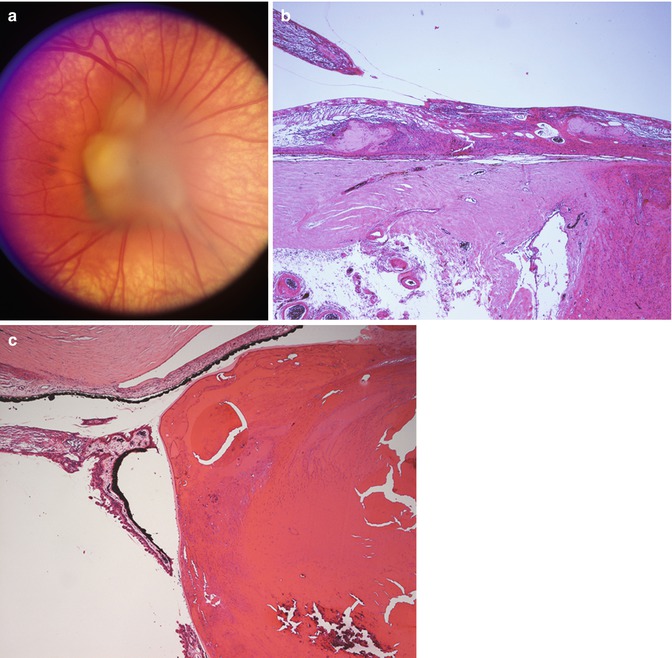
Fig. 9.2
Persistent fetal vasculature. (a) Clinical photograph showing gliosis and distortion of the retina surrounding the optic nerve. (b) Section through the nerve and peripapillary retina showing distortion of the retinal layers and gliosis. The persistent fetal vasculature is evident overlying the retina and is contiguous with the retinal tissue at other levels of section. (c) Anteriorly, the persistent fetal vasculature pulls the ciliary processes centripetally and is associated with cataractous changes in the lens
Another relatively common vascular anomaly that is of significance to the eye pathologist is Coats’ disease. In Coats’ disease, there are telangiectatic vessels within and on the surface of the retina. These vessels are prone to leakage, and the leakage of fluid and serum lipid can accumulate in the retina causing a mass that can mimic retinoblastoma (Fig. 9.3). This accumulation of fluid and lipid can be progressive over time, causing growth of the lesion, further confusing the clinical distinction between Coats’ disease and retinoblastoma. Eyes with Coats’ disease sometimes are enucleated due to concern that the intraocular changes could be retinoblastoma.
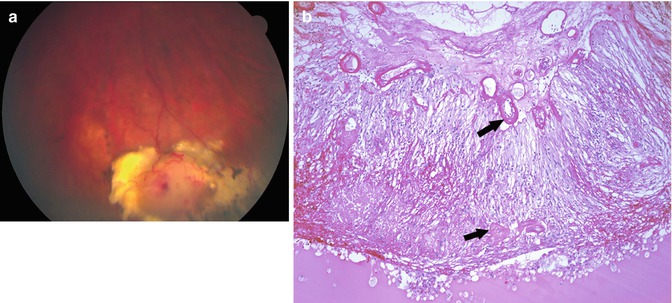
Fig. 9.3
Coats’ disease. (a) Clinical photograph showing white to yellow lesion in the retinal periphery with prominent vasculature. (b) Section showing telangiectatic vessels (arrows) within the retina and on the retinal surface. The retina is detached. There are lipid-laden macrophages in the subretinal space
There is a vast literature on the clinical features of the inherited retinal degenerations, and a complete summary is beyond the scope of this chapter. However, the clinical features of three of the diseases listed in Table 9.2 will suffice to provide correlative material for the histopathological section that follows. Retinitis pigmentosa is an inherited retinal degeneration characterized by initial difficulty in seeing at night (nyctalopia) and loss of peripheral vision. With progression, central vision may become affected as well. Retinitis pigmentosa is a heterogeneous disorder, with close to 200 individual causative mutations having been identified. It can be inherited as an X-linked recessive, autosomal recessive, or autosomal dominant trait. On examination, the fundus appearance is characterized by prominent RPE hyperplasia, with the pigmented cells giving a “bone spicule” appearance due to the predilection of the hyperplastic RPE cells to accumulate around retinal vessels (Fig. 9.4).
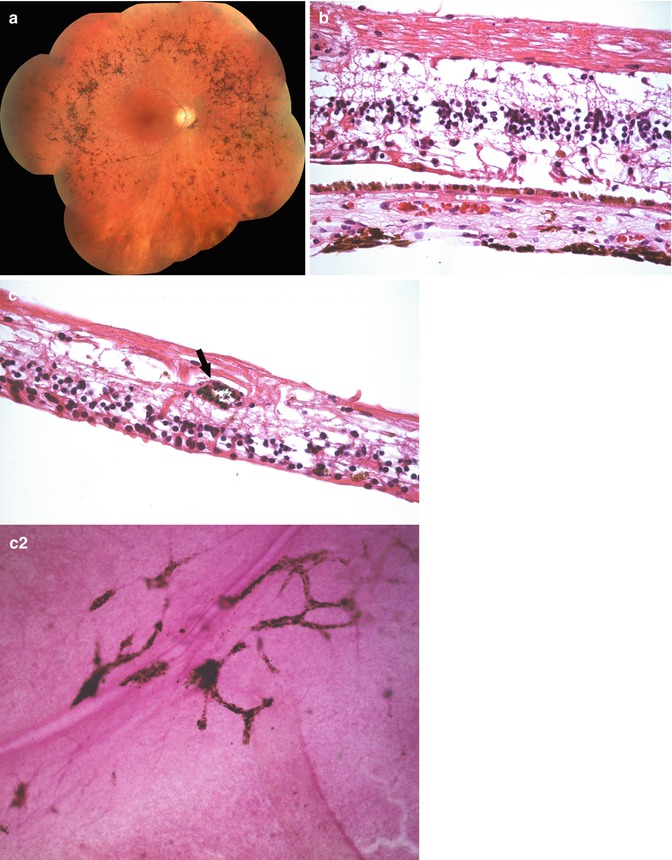
Fig. 9.4
Retinitis pigmentosa. (a) Clinical photograph showing marked RPE hyperplasia and atrophy in the peripheral retina. (b) Histopathology reveals there is marked loss of the photoreceptor layers. The inner retina has some alteration in the normal lamellar architecture consistent with some remodeling occurring due to the loss of photoreceptors. (c) The hyperplastic RPE has migrated into the retina and surrounds retinal vessels (arrow) as shown in the (c2) retinal whole mount and cross section
Gyrate atrophy is another inherited retinal degeneration, which is due to genetic defects affecting ornithine aminotransferase. Gyrate atrophy is an autosomal recessive disorder and is prototypical of the inherited retinal degenerations that are characterized by retinal atrophy as well as RPE atrophy (Fig. 9.5). In gyrate atrophy, patients are symptomatic due to their loss of peripheral visual field.
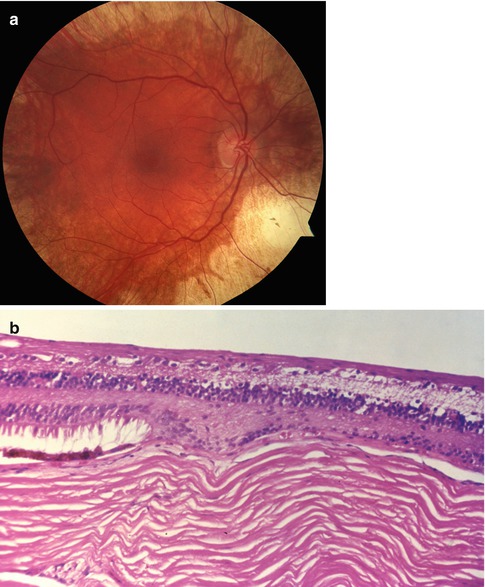
Fig. 9.5
Gyrate atrophy. (a) Clinical photograph showing abrupt transition from small area of relatively normal retina in macula and surrounding nerve to area of marked atrophy of the choroid, RPE, and retina. (b) Section in the transition zone showing complete loss of the RPE and anterior choroid with preservation of the inner retinal layers
Sorsby’s macular dystrophy is an autosomal recessive trait characterized by extensive changes in the extracellular matrix due to an abnormality of a tissue inhibitor of metalloproteinase (TIMP-3). Patients with Sorsby’s disease have prominent deposits of extracellular material beneath the retina. This material and associated damage to Bruch’s membrane are associated with the growth of abnormal blood vessels beneath the retina – so-called choroidal neovascularization. The growth of blood vessels in this location leads to scarring with associated severe decrease in vision (Fig. 9.6).
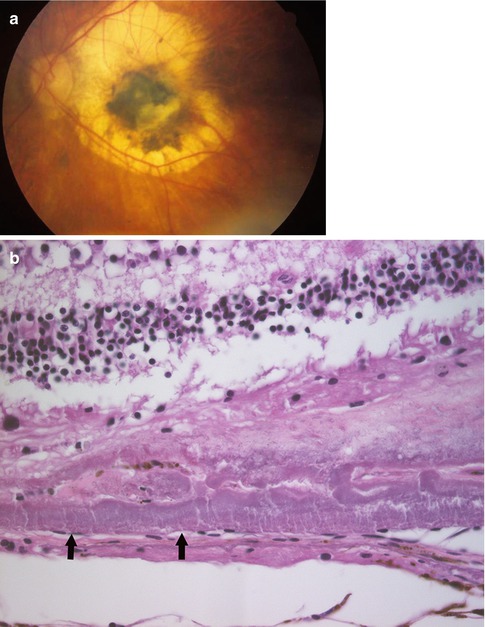
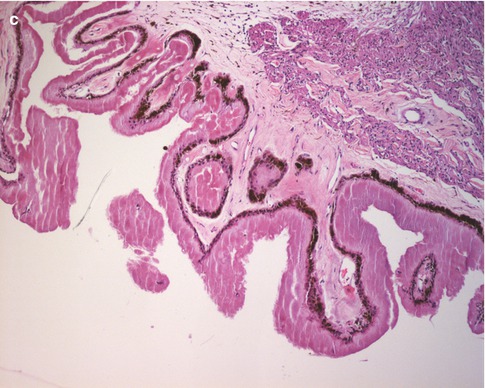
Fig. 9.6
Sorsby’s macular dystrophy. (a) Clinical photograph showing the macular changes of subretinal exudate and RPE hyperplasia secondary to subretinal neovascularization associated with this condition. (b) Histological sections demonstrate a marked increase in extracellular matrix both in the area of Bruch’s membrane. (c) Similar extracellular material associated with the nonpigmented epithelium of the ciliary body
In Stargardt disease, any of the three stereotypical tissue responses can occur. Some patients maintain relatively good vision and only manifest some subretinal deposits. Others have moderate disease that manifests as focal loss of photoreceptors and atrophy of the underlying RPE. And the most severely affected patients have diffuse loss of photoreceptors associated with RPE hyperplasia and migration into the retina.
Histopathology
Of the structural and developmental anomalies, the conditions of greatest relevance to a general pathologist are persistent fetal vasculature and Coats’ disease. The histopathology of persistent fetal vasculature is variable, but in the more extensive cases, there is persistence of hyaloid vessels arising from the optic nerve and extending anteriorly to the posterior lens and ciliary body. The tissue associated with the vessels can contain myofibroblasts, and traction on adjacent tissues can lead to traction detachment of the peripapillary retina posteriorly and central dragging of the ciliary processes in the anterior portion of the eye. Contact with the posterior lens capsule is associated with cataract formation (Fig. 9.2).
In Coats’ disease, there are telangiectatic vessels in the retina. In enucleated eyes, there is generally a total exudative retinal detachment present. In the subretinal space, there are often macrophages that have “foamy” lipid-containing cytoplasm. Cholesterol clefts may be present in the subretinal space consistent with the prior presence of abundant cholesterol (Fig. 9.3).
Histopathological study of the eyes from patients with retinitis pigmentosa reveals there is marked loss of the photoreceptor layer [6]. This is most severe peripherally, but in advanced cases, there can be loss of photoreceptors in the fovea as well. The RPE becomes hyperplastic, and there is migration of pigment-laden RPE cells to a position surrounding the retinal vasculature (Fig. 9.4). In addition to the striking changes at the level of the photoreceptors and RPE, there are changes that occur in the inner retina. The normal inner retinal circuitry is not maintained, and there is sprouting of neurites and gliosis. Both of these latter changes may limit future therapeutic options as the inner retinal changes may compromise re-establishment of a functioning visual pathway.
In patients with gyrate atrophy [7], there is loss of the photoreceptor layer, but unlike retinitis pigmentosa, the RPE does not become hyperplastic. In fact, the RPE in the areas of photoreceptor degeneration is completely absent. Where the photoreceptors are preserved, they have a remarkably normal appearance (Fig. 9.5).
Sorsby’s macular dystrophy [8] is characterized by prominent deposits of eosinophilic material beneath the RPE and the ciliary epithelium (Fig. 9.6). These deposits are associated with the ingrowth of blood vessels from the underlying choroid. Leakage of fluid from this neovascularization results in hyperplasia of the RPE and atrophy of the overlying photoreceptors.
Prognosis and Predictive Factors
Congenital structural abnormalities are only progressive on the basis of secondary changes that occur as a result of the anomaly. For example, with Coats’ disease, progressive leakage from the telangiectatic vessels can lead to retinal detachment. In addition, this leakage and secondary gliosis can lead to an intraocular mass that masquerades as retinoblastoma. Treatment of the abnormal blood vessels with laser or cryotherapy can prevent either retinal detachment or the development of a large mass. However, frequently the presence of the abnormal vessels is not known early enough to permit treatment.
The prognosis for the inherited retinal degeneration varies based on the disease. In general, diseases characterized by diffuse photoreceptor atrophy and RPE hyperplasia have a poor prognosis for vision. An intermediate prognosis is present for those diseases with regional photoreceptor atrophy associated with RPE atrophy. Those diseases with genetic abnormalities primarily affecting the extracellular space have a variable prognosis based on the secondary complications that may ensue. Mild Stargardt disease has a fairly good prognosis whereas Sorsby’s macular dystrophy is associated with severe vision central vision loss.
9.4 Retinal Inflammatory Conditions
Definition
Diseases that have a predominant inflammatory etiology can be further classified into those that are due to an identifiable infectious agent (Table 9.3) and those that are noninfectious (Table 9.4). This classification is of benefit both from a clinical standpoint in which treatment will be markedly affected by identifying an infectious agent; but, also the pathology of infectious inflammatory conditions is quite distinct from the noninfectious inflammatory disorders.
Table 9.3
Infectious retinal inflammatory conditions
Viral |
Cytomegalovirus |
Herpes simplex |
Herpes zoster |
Bacterial |
Exogenous |
Postsurgical |
Traumatic |
Endogenous |
Fungal |
Exogenous |
Postsurgical |
Traumatic |
Endogenous |
Protozoal (toxoplasmosis, etc.) |
Table 9.4
Noninfectious retinal inflammatory conditions
Autoimmune retinopathy |
Cancer-associated retinopathy (CAR) |
Melanoma-associated retinopathy (MAR) |
Acute zonal occult outer retinopathy (AZOOR) |
Multiple evanescent white dot syndrome (MEWDS) |
Adamantiades–Behçet’s disease |
Eales’ disease |
Epidemiology
Infectious retinitis is relatively common. The most common cause for infectious retinitis is bacterial infection following cataract surgery. This type of infection occurs in 0.03–0.16 % of cases [9, 10]. Bacterial or fungal retinal infections also occur without prior surgery, so-called endogenous endophthalmitis. This latter type of infection occurs most commonly in patients with indwelling central lines but may also be the result of systemic immunosuppression as is seen in post organ transplant patients. Endogenous bacterial endophthalmitis comprises 2–8 % of bacterial endophthalmitis [11]. Viral retinal infections are most common in immunosuppressed patients, most typically HIV-infected patients with low CD4 counts. Viral retinal infections also occur as reactivation of herpes zoster. Finally, toxoplasmosis is a frequent cause of infectious retinitis. Toxoplasmosis can be an acquired infection or can be the result of reactivation of a congenitally acquired infection.
There are certain epidemiologic associations with noninfectious retinitis as well. Notably, autoimmune retinopathy occurs in association with systemic malignancy, and Adamantiades–Behçet’s disease is associated with the HLA B51 haplotype.
Etiology
The etiology of the conditions resulting from infectious organisms is straightforward. The various infectious agents gain access to the eye either as the result of introduction during surgery (postsurgical endophthalmitis), as the result of injury, or due to hematogenous dissemination. In the case of toxoplasmic retinitis, the initial retinal infection may have occurred in utero due to transplacental infection with toxoplasmosis. Quiescent, postinfectious scars may harbor encysted toxoplasmic organisms which reactivate creating acute disease in children or adults.
The etiology of autoimmune retinopathy has been speculated to be similar to other paraneoplastic disorders: a remote malignancy elaborates an antigenic substance that is sufficiently similar to a retinal antigen that the resulting antibody and immunologic reaction results in damage to the retina. In the case of AZOOR and MEWDS, the autoimmune response is hypothesized to be triggered by a previous viral infection.
Clinical Features
Postsurgical infections present as intense acute inflammation in the aqueous and vitreous. The presence of the intense neutrophil infiltrate obscures vision resulting in symptoms of decreased vision, photosensitivity, and pain. Most cases of postsurgical endophthalmitis will be diagnosed on the basis of culture of aqueous and vitreous fluid. Typically, intravitreal injections of antibiotics or antifungals will be administered immediately following acquisition of fluid for culture. Injections of this type are usually effective in eradicating the infection, but in some cases, the extent of retinal damage or secondary changes will result in enucleation of a painful globe. This is more likely to occur in cases of infections with more aggressive bacteria, such as pneumococcus or Gram-negative bacteria (Fig. 9.7). Posttraumatic infections, particularly if Bacteroides is the infecting agent, are also likely to result in severe retinal injury that may lead to enucleation of the globe.
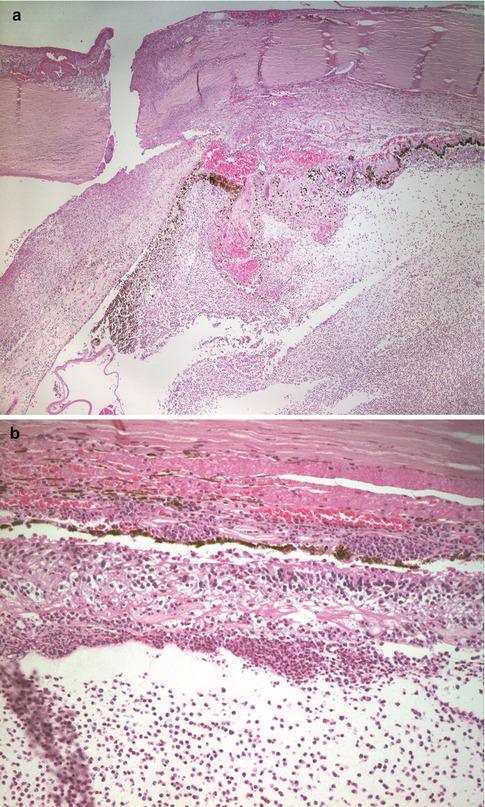
Fig. 9.7
Postoperative endophthalmitis. (a) The unhealed limbal wound is evident. There is marked acute inflammation in the anterior and posterior chambers. (b) There is marked acute inflammation involving the vitreous, with infiltration of the retina and choroid
Endogenous endophthalmitis also presents as decreased vision due to inflammation-induced loss of clarity of the optical media or due to direct infection of the central retina. Examination discloses a white infiltrate in the retina with overlying inflammation (Fig. 9.8). The patient typically has an indwelling central line or is immunocompromised. CMV infection of the retina is a common finding in patients infected with HIV who have CD4 counts below 50 per ml. CMV retinitis has a distinctive clinical appearance with the retina having dense whitening associated with intraretinal hemorrhage (Fig. 9.9). Strikingly, there is generally very little vitreous inflammation associated with the retinal infection. This aids in distinguishing CMV infection from acute retinal necrosis (see below) and toxoplasmosis. As the infection destroys the retina, the retinal whitening disappears and is replaced by markedly thinned retina with pigmentation due to RPE hyperplasia. The infection proceeds centripetally from its initial focus so that in more long-standing infections there are areas of thinned, variably pigmented retina surrounded by a border of whitened retina representing the location of the active infection.
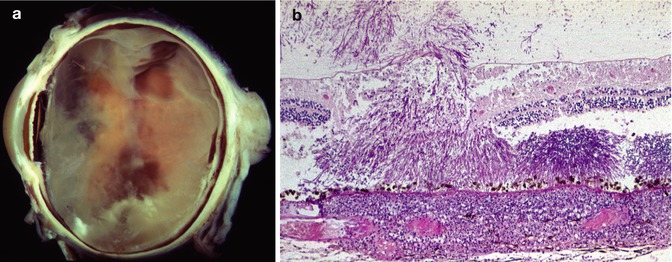
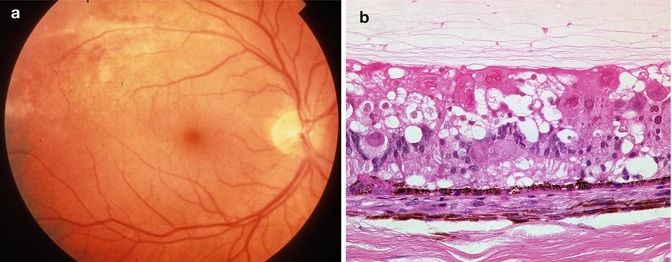
Fig. 9.8
Endogenous fungal aspergillus endophthalmitis. (a) Gross photograph showing intraretinal hemorrhages and retinal whitening in the areas of fungal infection. There is also overlying vitreous opacity. (b) There are fungal aspergillus hyphae throughout the retina and extending into the vitreous. There is a prominent inflammatory in filtrate in the choroid
Fig. 9.9
CMV retinitis. (a) Clinical photograph. There is retinal whitening and intraretinal hemorrhage along the superior arcade. (b) Section through an area of CMV-retinitis reveal prominent cytomegalic cells with intranuclear and intracytoplasmic inclusions, as well as atrophy and necrosis in the inner and outer retinal layers
Acute retinal necrosis (ARN) describes a clinical presentation of marked retinal whitening due to necrosis (Fig. 9.10). Typically the area of involvement begins peripherally and moves centrally, although the opposite pattern has been reported (this latter pattern is referred to as progressive outer retinal necrosis, PORN). In contrast to CMV retinitis, ARN occurs in immunocompetent patients and is associated with prominent intraocular inflammation. Acute retinal necrosis may be due to herpes zoster or herpes simplex.
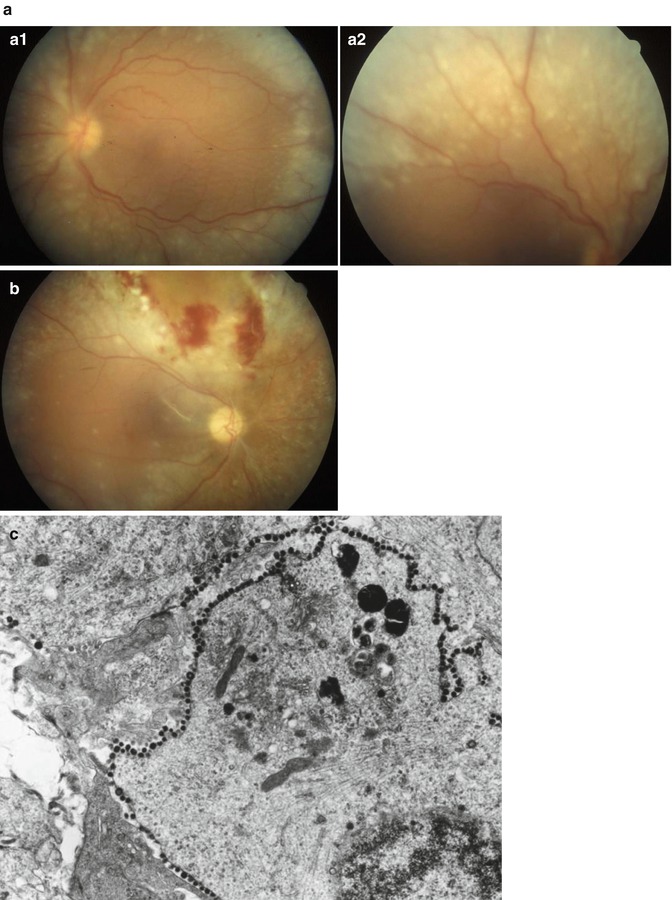
Fig. 9.10
Acute retinal necrosis. (a) Clinical photographs showing bilateral retinal whitening outside of the area of the retinal arcades. (a1) Left eye, (a2) right eye. (b) Clinical photograph showing retinal biopsy site, with mild retinal hemorrhage at the margin of the area of biopsy. (c) Electron microscopy of the biopsy shows numerous viral particles decorating the periphery of a retinal cell
Toxoplasmic retinitis also presents with decreased vision due to intraocular inflammation or due to direct involvement of the central retina with the infection (Fig. 9.11). The white retinal infiltrate is classically described as a “headlight” in the “fog” of vitreous inflammation. If the ocular media are sufficiently clear, it is possible to see that the area of active infection may be adjacent to an old scar. This latter feature is a strong indication that the infection is due to toxoplasmosis.
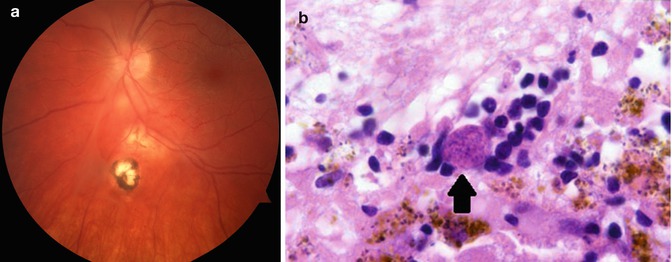
Fig. 9.11
Toxoplasmic retinochoroiditis. (a) Clinical photograph showing retinal scars at different stages of evolution. The most inferior lesion is an old pigmented scar. Superior to this there is a more recent area of retinal involvement, and inferior to the optic nerve there is an area of active retinitis. (b) Section through retina of a patient with toxoplasmosis. The retina is necrotic, and there are numerous encysted toxoplasmic organisms evident (arrow)
CMV, H. zoster, H. simplex, and toxoplasmosis may be diagnosed by PCR of vitreous or aqueous samples. In some cases in which the diagnosis is not established by PCR, retinal biopsy may be performed to try to confirm a diagnosis.
As indicated in Table 9.4, there are several noninfectious entities that may lead to retinal inflammation. There are several distinct clinical entities that are hypothesized to be a form of retinal autoimmunity: cancer-associated retinopathy (CAR), melanoma-associated retinopathy (MAR), acute zonal occult outer retinopathy (AZOOR), and multiple evanescent white dot syndrome (MEWDS).
The clinical manifestations of CAR are typically subacute onset and progressive loss of vision, nyctalopia, measurable visual field loss, and diminished electroretinogram. Some patients will also have floaters and photophobia. The vision loss may progress to very severe visual impairment. At the time of onset of visual symptoms, patients may or may not have a history of prior malignancy. CAR is most often associated with lung cancer but has also been described in patients with carcinoma of the colon, uterus, cervix, breast, and prostate. The clinical manifestations of MAR are nyctalopia, pigmentary changes in the retina and vitiligo. Patients with MAR have clinical and electrophysiologic findings similar to patients with congenital stationary night blindness. MAR patients are likely to have already been diagnosed with cutaneous melanoma prior to the onset of their visual symptoms.
AZOOR and MEWDS are clinically defined entities that occur more commonly in women. AZOOR is characterized by patients that present with the abrupt onset of a blind spot and photopsias. The fundus initially appears normal but with time an area of retinal atrophy and RPE hyperplasia develops. There is no sign of inflammation.
MEWDS patients often describe a flulike illness preceding their symptoms of decreased vision and photopsias. Examination discloses a granular appearance to the fovea and faint, transient white opacities at the level of the retinal pigment epithelium and deep retina.
Adamantiades–Behçet’s disease is a systemic vasculitis that has prominent ophthalmologic features. In addition to the orogenital ulcers present in Adamantiades–Behçet’s disease, there is prominent intraocular inflammation with occlusive retinal vasculitis.
Eales’ disease is characterized by the presence of a peripheral perivasculitis involving retinal veins. Venous occlusions lead to non-perfusion and peripheral retinal neovascularization. Vitreous hemorrhages and traction retinal detachments may develop secondary to the neovascularization. The disease is typically bilateral, but there may be a considerable time interval before involvement of the second eye.
Histopathology
In acute bacterial infections (endophthalmitis) resulting from surgical procedures, there is a marked infiltrate of neutrophils in the choroid, retina, and vitreous. The bacteria causing the infection were generally introduced into the eye at the time of surgery, so there is typically no bacterial infiltrate in the eye wall. Most cases of endophthalmitis that come to the pathology laboratory will have been treated aggressively with intravitreal antibiotics, so it is uncommon to see bacterial organisms even in severe cases of endophthalmitis. Edema and hemorrhage in the retina are common in longer-standing infections.
With endogenous fungal endophthalmitis, fungus reaches the eye through the bloodstream, and infection generally begins in the choroid. However, extension of the infection to the retina and vitreous is typical. Both yeasts and hyphae may be evident within the area of infection. Lesions may have the histopathological features of an abscess with central necrosis surrounded by acute inflammation, with a perimeter of chronic and granulomatous inflammation.
Retinal infection with CMV leads to full-thickness destruction of the retina and with healing there is a thin fibroglial scar replacing the retina. In active infections, there are intranuclear and intracytoplasmic eosinophilic inclusions within large neurons (cytomegalic cells).
Similarly with acute retinal necrosis from either herpes simplex or herpes zoster, there is necrosis of the full thickness of the retina. Occasional intranuclear inclusions may be present. There is prominent retinal vasculitis and diffuse inflammation in the vitreous and uvea.
In toxoplasmic retinitis, the infection or reactivation of infection begins in the retina. There is necrosis of the retina with surrounding acute inflammation. The inflammatory response will extend into the vitreous and to the adjacent choroid. Cysts containing the Toxoplasma organism are often evident. Free organisms may also be seen.
The histopathology of CAR is strikingly different than that of the infectious retinal inflammatory conditions (Fig. 9.12). In CAR, there is no necrosis and cell death appears to be the result of apoptosis. There is marked and relatively selective loss of photoreceptor cells. These histopathological findings are consistent with retinal autoantibody-induced damage to the photoreceptor cells.
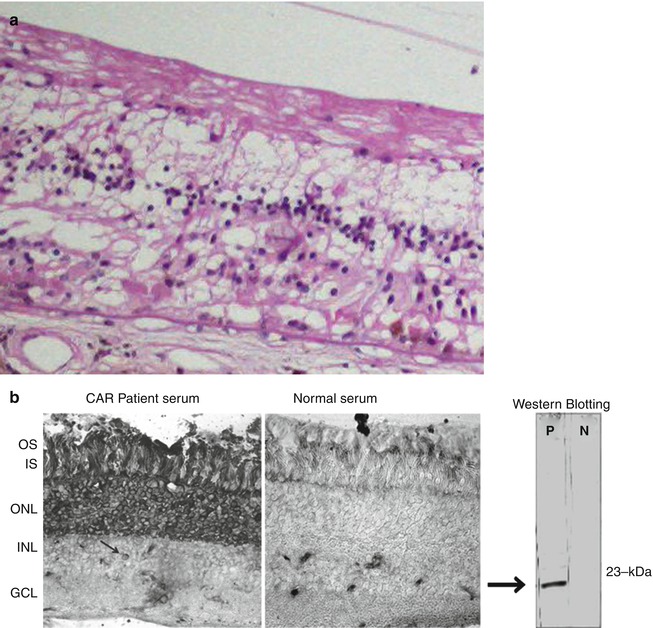
Fig. 9.12
Cancer-associated retinopathy. (a) There is complete loss of the photoreceptor layer and partial loss of the inner nuclear layer; notably there is no inflammation. The RPE is partially absent. In other areas the RPE is hyperplastic with some migration into the retina. (b) Immunohistochemistry and Western blot of serum from a patient with CAR. The Western blot shows there is an antibody in the patient’s serum that reacts with a 23-kDa protein (recoverin). Immunoperoxidase staining of normal retina shows that when the patient’s serum is used as a primary antibody (left panel), there is prominent labeling of the photoreceptor nuclei, inner and outer segments
In MAR, again, there is minimal to no inflammatory cell infiltrate. The cell loss in MAR is at the level of the inner nuclear layer and in particular appears to involve primarily ON bipolar cells [12]. There is also narrowing of the outer plexiform layer.
The histopathology of Behçet and Eales’ disease is similar and is characterized by a vaso-occlusive vasculitis. Histological findings of retinal ischemia: cotton wool spots, retinal hemorrhage, and retinal thinning are generally present. Acute and chronic inflammation within the iris and ciliary body are also present.
Prognosis and Predictive Factors
The prognosis for bacterial endophthalmitis is based on the infecting organism and the time between onset of the infection and treatment. As most bacterial endophthalmitis is postsurgical and is the result of Staphylococcus epidermidis, a relatively benign bacteria, the outcome in these patients is generally good. In the Endophthalmitis Vitrectomy Study, more than 50 % of patients achieved a visual acuity of 20/40 or better at 9 months following treatment. Seventy-five percent of patients achieved a vision of 20/100 or better [13].
The visual outcome for endogenous endophthalmitis is also variable, depending primarily on the organism causing infection, the immune status of the patient, and the location of the retinal involvement. Mild Candida endophthalmitis, diagnosed early and treated, has an excellent prognosis. Bacterial endogenous endophthalmitis [14] and Aspergillus endophthalmitis in an organ transplant patient have a poor prognosis for vision. In addition, endogenous Aspergillus endophthalmitis is almost certainly associated with disseminated Aspergillus, and the prognosis for patient survival is poor [15].
CMV retinitis is very amenable to treatment, either by improving the patient’s immune status, use of systemic or intravitreal antiviral agents or both. HIV-infected patients that develop CMV retinitis are generally treated with systemic and/or intravitreal antiviral agents but can often be weaned from these if their CD4 counts respond to highly active antiretroviral therapy (HAART).
Of the patients with noninfectious retinal inflammatory conditions, patients with MEWDS have an excellent visual prognosis and generally recover normal vision with no treatment. AZOOR patients may show progression but usually achieve an inactive status over the course of 1 year. The involvement is sectoral, and central vision is usually spared.
Patients with ARN or PORN can also be treated with antiviral therapy. These patients’ visual prognosis depends on the extent to which the central retina has been damaged from direct viral infection and whether they develop a secondary retinal detachment. Similarly, toxoplasmosis patients will generally maintain good vision as long as the central retina is not directly damaged by the initial or recurrent bouts of active infection.
Cancer-associated retinopathy has been treated with immunosuppression. There are anecdotal reports of stabilization and improvement in vision, but no large series are available to confirm a specific treatment or prognosis.
9.5 Retinal Injury
Definition
Retinal injury can be defined as any perturbation that disrupts the cellular constituents or extracellular matrix of the retina sufficiently to cause transient or permanent alterations in retinal function, physiology, or structure. Defined in this fashion, retinal injury is a broad term that can encompass changes in the retina that occur secondary to a variety of processes. Permanent changes in the cellular makeup of the retina occur secondary to apoptosis, necrosis or autophagy. Permanent changes might also occur due to changes that are largely in the extracellular matrix, e.g., following vitreous detachment. Apoptosis is a process of programmed cell death. Apoptosis can be elicited by several mechanisms of injury, including inherited retinal diseases or metabolic disease. Characteristically, it occurs in the absence of inflammation. Necrosis occurs more typically with mechanical trauma or infectious injury mechanisms. Autophagy is a process of degradation of dysfunctional cellular components. In extreme situations it may be viewed as another form of programmed cell death. There are, of course, types of injury that elicit both apoptosis and necrosis, for example, ischemic injury. As ischemic injury and retinal degeneration are covered as separate topics, this section will deal with retinal injury as a result of direct and indirect mechanical and thermal injury to the retina.
Epidemiology
Mechanical eye injury is very common. It is estimated that over two million eye injuries occur each year in the USA [16]. Predisposing factors for injury relate to occupation, age, and sporting activities. Males are much more likely to sustain eye trauma than females. A special form of eye trauma is nonaccidental trauma seen as a result of child abuse. The constellation of mechanical eye injuries in this setting is generally referred to as “shaken baby syndrome” and occurs primarily in children under the age of 1.
A subcategory of mechanical eye trauma is surgical trauma. In the case of surgical trauma, injury to eye tissue is purposeful, in an effort to improve a disease state. Surgical trauma to the retina occurs as a result of laser treatment and with surgical incisions in the retina. Laser-induced injury to the retina is very common, while surgical incisions of the retina are less common. The pathology of both types of injury is relevant to the interpretation of tissue received in the pathology laboratory.
Clinical Features
Blunt trauma to the retina induces multiple types of retinal injury. Injury to the retina occurs as a result of direct trauma and from counter coup injury due to vitreous perturbations from deformation of the globe. Commotio retina (Fig. 9.13a) is a term that describes the clinical appearance of the retina with blunt injury. This retinal whitening is due to shearing of the photoreceptor outer segments [17]. With more severe trauma, the injury can be more extensive, and full-thickness necrosis of the retina may occur and is called “sclopetaria” (Fig. 9.13b, c). Blunt trauma may also result in retinal injury in the form or retinal tears, dialysis, or macular hole. Retinal and vitreous hemorrhage is a frequent accompaniment of this type of injury. Retinal detachment is a common sequela following retinal tear.
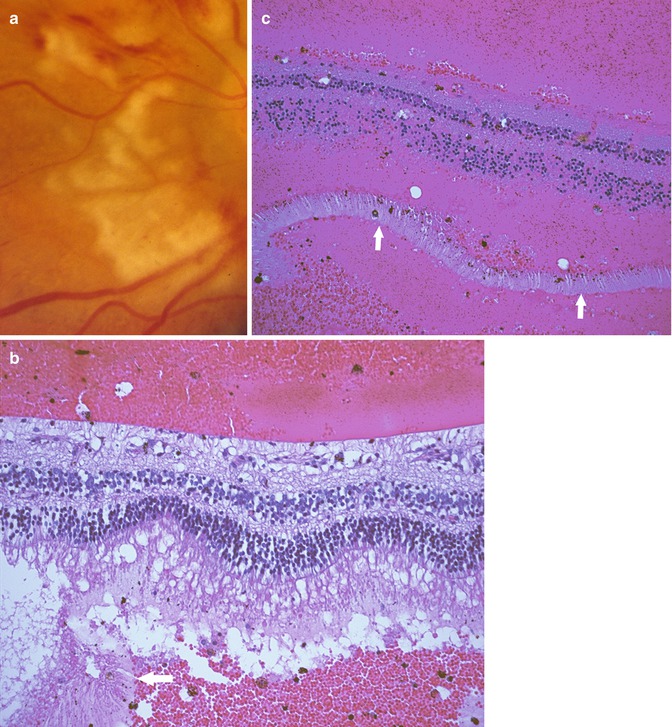
Fig. 9.13
(a) Clinical photograph showing severe blunt retinal injury resulting in full-thickness necrosis of the retina, so-called retinal sclopetaria. (b) Section of adjacent retina shows separation of the outer segments (arrow), but the remaining retina is relatively intact. (c) Histopathology of full-thickness retinal trauma. There is necrosis of the full thickness of the retina. The outer segments of the photoreceptors (arrows) are separated from the overlying retina. The retinal nuclei are hyperchromatic
As mentioned above, a special form of blunt injury occurs with the nonaccidental trauma of “shaken baby syndrome.” With this injury, there are numerous intraretinal hemorrhages, generally present in both the anterior and posterior retina. As the retinal injuries are typically accompanied by head trauma resulting in subarachnoid hemorrhage, there is often prominent optic nerve head edema present [18].
Penetrating or perforating injuries of the eye may result in lacerations or tears of the retina. Such injuries are often complicated by a later development of retinal detachment due to the cellular proliferation that occurs as part of the wound healing process [19].
Laser treatment to the retina induces thermal injury (Fig. 9.14). Laser treatment is done for three principal reasons: to create an adhesion between the retina and the underlying RPE and choroid, to indirectly induce regression of retinal vessels and retinal neovascularization, and to directly destroy a lesion.
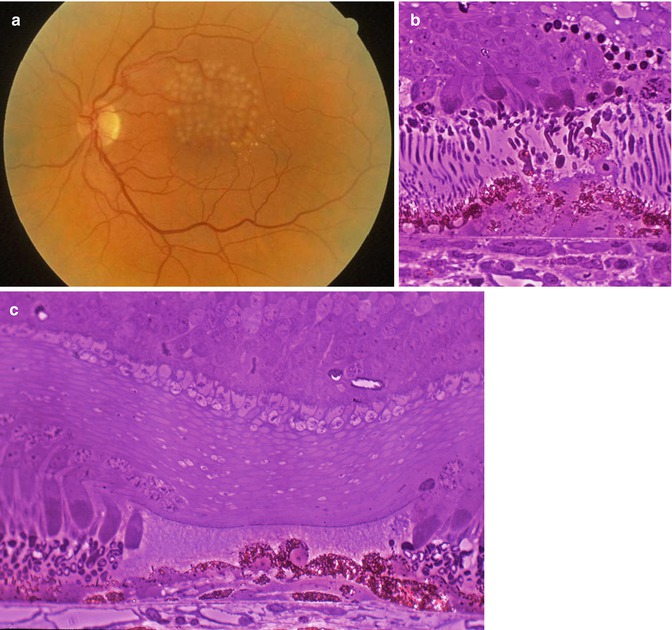
Fig. 9.14
(a) Clinical photograph of laser photocoagulation burns in the retina. (b) Acute laser burn showing necrosis of the photoreceptor layer and RPE cells of the retina. (c) Healed laser burn showing the focal defect in the photoreceptor layer that is filled primarily with extracellular matrix. The RPE cell monolayer has reformed, but there is some RPE hyperplasia in the area of the laser treatment
Histopathology
Age-related degeneration of the vitreous or blunt trauma can result in vitreous traction on the retina sufficient to cause a retinal tear (Fig. 9.15). The retina contains very little collagenous extracellular matrix, so the structural integrity of the retina is principally derived from the retinal vasculature. The local tissue response to a retinal tear consists of loss of photoreceptor cells in the area of the tear and focal gliosis; but more importantly, a retinal tear predisposes to a retinal detachment. Retinal detachment results in marked changes within the retina and the underlying RPE. Changes begin almost immediately following detachment. Early changes include transition of the Müller cell from a cell that is supporting neuronal function to a cell forming glial scars on the anterior and posterior surface of the retina. Normally, the RPE cell is mitotically inactive, but following retinal detachment, RPE cells begin to “bud off” from the RPE monolayer and migrate into the subretinal and vitreous spaces. The photoreceptor outer segments degenerate. Approximately 3 days to one week following detachment apoptosis of the photoreceptor cells begins. With chronic detachment, the retina loses its lamellar architecture. The neuronal cell populations are absent, and the retina consists of primarily glial tissue. There can be membranes on the anterior and posterior surface of the retina, composed primarily of RPE cells and glial cells. The clinical term used to refer to these membranes is proliferative vitreoretinopathy. The constellation of retinal changes occurring with retinal detachment has been well characterized in experimental studies of retinal detachment and in human postmortem studies [20, 21].
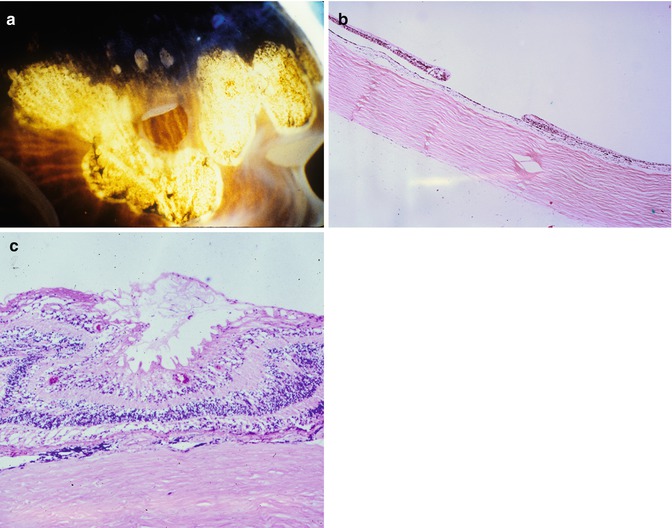
Fig. 9.15
(a) Gross photograph of a retinal tear, surrounded by cryotherapy scars. (b) Histopathology showing retinal defect corresponding to the tear. Note there is mild glial proliferation at the margin of the tear. (c) Proliferative vitreoretinopathy characterized by cellular proliferation on the surface of the retina. The surface of the retina is wrinkled due to the presence of a glial membrane on the vitreous surface of the retina. There is marked loss of the photoreceptor layer, and there is a thin glial membrane beneath the retina
Penetrating or perforating injuries of the retina have some similarities to retinal tears but are much more likely to result in transection of the vessels within the retina. When the retina is lacerated, there is no capacity for healing of the defect. So, if no retinal detachment ensues, the margin of the retina heals by glial cell proliferation with the development of a chorioretinal adhesion (Fig. 9.16).
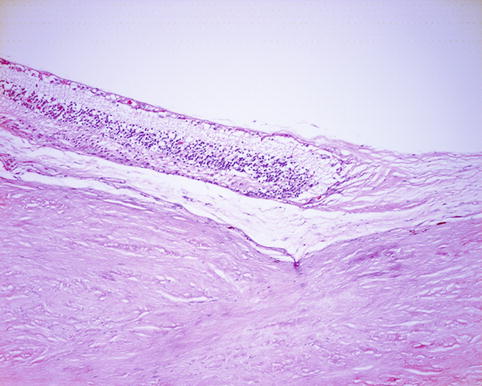
Fig. 9.16
Healed retinal laceration site. There is abrupt termination of the retina with the defect filled with fibrous tissue
Thermal laser photocoagulation is a very common type of retinal injury. Most lasers used in treating retinal disease or during retinal surgery have a wavelength that leads to energy absorption by hemorrhage or melanin pigment (pigment within the RPE or choroid). The extent of the retinal damage will be determined by the power of the laser setting and the amount of hemorrhage and pigment in the path of the laser beam. With laser treatment, the type of injury is thermal necrosis. Typically, the power of the laser is adjusted to obtain necrosis of the outer retina and RPE while retaining an intact inner retina. Figure 9.14 illustrates the very focal RPE and outer retinal injury that occurs with laser photocoagulation. With healing, the RPE adjacent to the injury site divides and reforms the RPE monolayer. There is no healing of the photoreceptor layer, and the absence of cells in the photoreceptor layer is filled with extracellular matrix or glial cell processes. In addition to the loss in the retina and the RPE changes, there is loss of the choriocapillaris in the area of laser treatment. This does not appear to reform following photocoagulation.
Prognosis and Predictive Factors
A common clinical endpoint for many types of retinal injury is retinal detachment. For retinal detachment, the injury and wound healing factor that is most predictive of prognosis are the presence or absence of proliferative vitreoretinopathy. If severe membranes are present on the anterior or posterior surface of the retina, it is much more likely that retinal detachment will be present and/or recur following surgical repair. In addition, glial cell membranes that extend over the posterior surface of the retina interfere with the re-establishment of photoreceptor–RPE contact that is essential for normal retinal function. Consequently, retinas with posterior membranes are unlikely to have as much visual acuity recovery even with successful retinal reattachment. Recovery of vision will also depend on the preservation of the photoreceptors. As noted above, photoreceptor apoptosis occurs 3–7 days following detachment, so prognosis will depend on the duration of the detachment as well as wound healing factors.
9.6 Retinal Degenerations
9.6.1 Age-Related Macular Degeneration
Definition
Age-related macular degeneration (AMD) is a degenerative condition affecting the retina, Bruch’s membrane, and choroid. Early disease is characterized by focal and diffuse sub-RPE deposits, with or without pigment irregularity and later disease by associated choroidal neovascularization and/or atrophy of pigment epithelium and outer retina known as geographic atrophy.
Epidemiology
AMD is the most common cause of untreatable blindness in the elderly [22]. It is especially prevalent in industrial societies. It is becoming more apparent in Asia, but interestingly there appear to be subtle phenotypic differences in different populations. Particularly in Asians, there is, in addition, a variant of AMD known as polypoidal choroidal vasculopathy [23]. There appears to be relatively little AMD in Africa.
Etiology
Smoking is a well-established risk factor, and diet probably has a role to play with evidence to suggest a “healthy” diet is protective and a “Western” lipid-rich diet a risk. Sunlight exposure, perhaps surprisingly, has not been a consistent risk factor in epidemiological investigations of AMD. There are strong genetic risk factors with major influence from polymorphisms associated with complement factor H (CFH) and the ARMS2/HTRA1 locus on chromosome 10 [24]. The CFH findings, particularly when associated with the existence of other genetic risk factors in other genes linked to the control of complement activation, have moved the role of inflammation and the innate immune system center stage. Other inherited disorders, including Sorsby’s fundus dystrophy, have focused attention on abnormalities of the extracellular matrix and its turnover. Further genes with high-risk polymorphisms and gene products with functions linked to lipid metabolism align with the potential importance of diet.
Clinical Features
In early AMD, patients may be asymptomatic or complain of poor night vision. Loss of rod photoreceptors is an early feature [25]. As choroidal neovascularization develops, there may be loss or distortion of central vision. Visual loss will be sudden if there is subretinal or intravitreal bleeding.
On fundoscopy, in early disease the main finding is of macular drusen (focal sub-RPE deposits). If very small and relatively few in number, these are considered part of normal aging and carry little risk of progression to visual loss. If larger or associated with pigmentary changes, the risks of eventual visual impairment increase. The classification of early AMD has recently been revised [26] and linked to risk of progression. Diffuse deposits cannot be seen clinically as there are no contrast-rich edges. Choroidal neovascular membranes can be associated with hemorrhage, edema, and/or scarring. Geographic atrophy can develop with a variety of patterns best seen with autofluorescence fundus imaging. Curiously the fovea is often spared until late in the evolution of geographic atrophy. What may be regarded as variants of AMD exist. In polypoidal choroidal vasculopathy, ectatic-looking vessels in the choroid, best visualized with indocyanine green angiography, may leak or bleed with relatively massive subretinal hemorrhage. Reticular pseudodrusen are increasingly recognized as a clinical feature of AMD [27] associated with risk of progression to blinding disease and characterized by what appear to be ribbonlike deposits and lesions which, particularly on near-infrared imaging, look like small dark rings with a central dot.
Macroscopy
The macroscopic features are essentially those seen on fundoscopy although the clouding of the retina that occurs ex vivo makes the focal deposits harder to see.
Histopathology
The histopathology of AMD is complex, reflecting significant variation from case to case as well as evolution of changes with time. An additional consideration is how the pathology of AMD links to aging, so it is important to first describe what might be seen in the eye of an aged individual without AMD.
One common feature of aging and AMD is the presence of focal sub-RPE deposits, known as drusen. When these are relatively homogeneous with well-defined edges, they are known as hard drusen (Fig. 9.17), and a scattering of these, especially if small (<75 um) and extramacular, is a common finding in elderly eyes. Another common, indeed almost universal feature of the aging eye is thickening of Bruch’s membrane [28, 29]. This thickening is within the inner collagenous zone through to the outer collagenous zone and does not, typically, include a significant degree of basal laminar deposit formation (see below). The other finding that is not apparent in routine sections but can be visualized clinically with fundus autofluorescence imaging or under fluorescence microscopy (the fluorescein filter set works well) is the accumulation of lipofuscin in the retinal pigment epithelium [28]. Finally, in normal aging, there is loss of endothelium from the choriocapillaris [29]. This may not be apparent as the relatively solid connective tissue around the vessels remains intact, but if an endothelial cell marker is used, many of these spaces appear empty (see below).
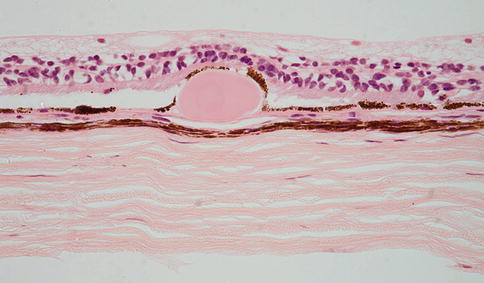
Fig. 9.17
Peripheral retina overlying a small, hard druse. The retinal pigment epithelium partially covers the druse. The choroid in this case is very thin
In early AMD there may be many hard drusen but also soft drusen (Fig. 9.18), which in contradistinction to hard drusen, often appear more heterogeneous, involve a greater extent of Bruch’s membrane and are of particular significance with regard to AMD and risk of disease progression to geographic atrophy or choroidal neovascularization. Sometimes they appear more apparent clinically than histologically, and in this instance they are probably at least in part pigment epithelial detachments (Fig. 9.19), a further well-recognized clinical feature of AMD. Important in considering their relation to the diffuse deposits discussed below, hard and soft drusen form external to the RPE basement membrane. Reticular pseudodrusen are an important risk factor for advanced disease, but the histopathological correlate of these phenomena is controversial and discussed below.
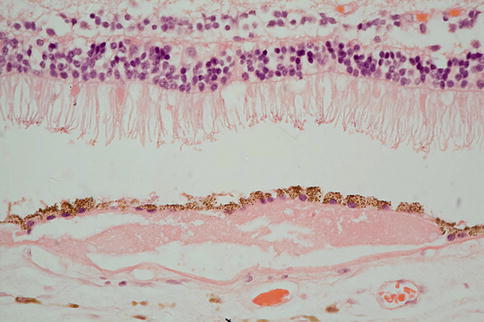
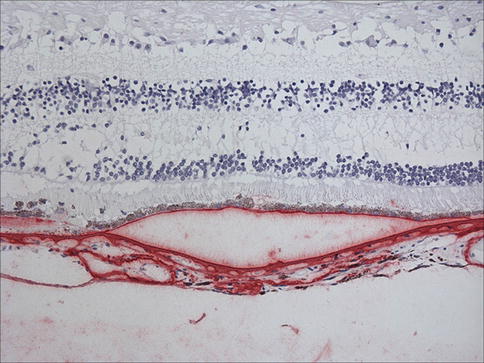
Fig. 9.18
There is neural retina at the top of the figure and reasonable numbers of rod outer segments remain. The neural retina is separated from RPE by an artifactual space. Beneath the RPE, there is amorphous eosinophilic material that would have probably appeared as a soft druse clinically
Fig. 9.19
Retina and choroid with immunostaining for vitronectin (red). Note that RPE is separated from Bruch’s membrane. This represents a pigment epithelial detachment
The classification of diffuse deposits has been confusing in the past, but there is now reasonable consensus that the protein-rich material that accumulates between the RPE and the RPE basement membrane should be called basal laminar deposit (Fig. 9.20) and the often more lipid-rich material external to the RPE basement membrane but internal to or involving the inner collagenous layer of Bruch’s membrane should be called basal linear deposit [30]. Basal laminar deposit often includes wide-spacing material (sometimes known as long-spacing collagen), which may also be seen at other levels within the Bruch’s membrane complex. Sometimes within H + E-stained sections, basal laminar deposit has a striated appearance (Fig. 9.21) with the striations oriented perpendicular to Bruch’s membrane. However, without electron microscopy, it is not possible to distinguish between basal linear deposit and basal laminar deposit. As discussed above, Bruch’s membrane complex thickens with age, at least in part due to small amounts of basal laminar and basal linear deposit, but the total increase in diffuse deposits is much greater in AMD. As a rule of thumb, if the thickening is readily appreciated in an H+E staining section, it is likely to be significant. The biochemistry of drusen and diffuse deposits is complex and includes complement components, coagulation factors, vitronectin (Fig. 9.19) [31], and TIMP-3 (Fig. 9.22).
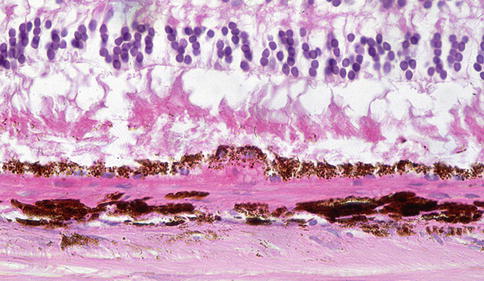
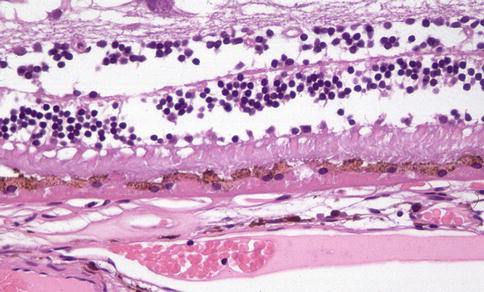
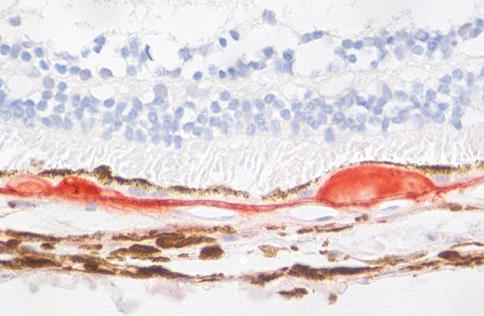
Fig. 9.20
Outer retina showing disorganization of photoreceptor outer segments. There is eosinophilic material beneath the RPE. It is not possible to distinguish on which side of the RPE basement membrane this is by light microscopy, but at least a component is likely to be internal to the basement membrane and hence basal laminar deposit
Fig. 9.21
There are poorly defined striations in the sub-RPE material. This is typical of basal lamina deposit although confirmation would require electron microscopy
Fig. 9.22
Sub-RPE deposit with immunoreactivity for TIMP-3 (red-brown)
Clinically, another feature of early AMD with adverse prognostic significance is pigmentary abnormality. The histopathology of this has not been well characterized. Early rod loss, an important clinical manifestation of early disease, is hard to detect subjectively in sections. Similarly, even though degeneration of the choriocapillaris is quite possibly of significance in terms of pathogenesis, it is difficult to appreciate in routine sections. With immunohistochemistry for an endothelial cell marker, the “gaps” in the sub-Bruch’s connective tissue become much more apparent (Fig. 9.23).
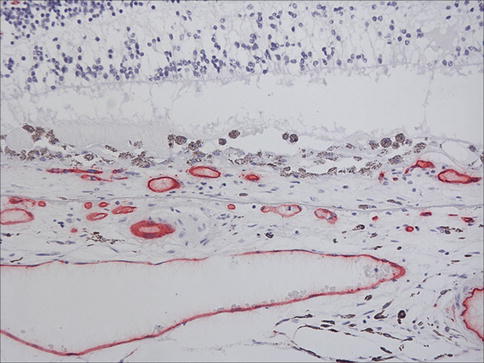
Fig. 9.23
Outer retina, choroidal neovascular membrane, and choroid from a case of age-related macular degeneration. Immunostaining for CD31 (red). Note vessels internal to Bruch’s membrane in the neovascular membrane and gaps under Bruch’s membrane where segments of choriocapillaris have been lost
Choroidal neovascularization (CNV) in AMD is generally sited at the posterior pole although it is important to note that peripheral CNV can occur and is sometimes encountered as an incidental finding. The number of breaches in Bruch’s membrane, which give the choroidal circulation access to the sub-RPE and subretinal spaces, is generally small [32], and no continuity between the choroid and the neovascular membrane is evident in most cases unless serial sections are examined. The neovascular membrane may, as described, be situated between the RPE and Bruch’s membrane (Fig. 9.24), the classical location for AMD-associated CNV, or between the neural retinal and RPE (Fig. 9.25) or indeed, both (Fig. 9.26). Sub-RPE membranes are sometimes referred to as type I membranes and subretinal membranes as type II. The surgical removal of sub-RPE membranes is generally not successful as the native RPE is removed as well, leaving the photoreceptors without any support leading to what is essentially geographic atrophy. The membranes often contain clumps of migrated RPE that may form apex-to-apex tubes and complexes, and the RPE will often depigment (Fig. 9.25). This can on occasion lead to histological misinterpretation as a retinal metastasis. The RPE may generate, within the membrane, material reminiscent of basal laminar deposit that can sometimes be extensive (Fig. 9.27). There is often a scattering of inflammatory cells (mainly lymphocytes and macrophages (Fig. 9.28) with rare mast cells), and a giant cell reaction to damaged Bruch’s membrane or basal laminar material (Fig. 9.29) is occasionally seen.
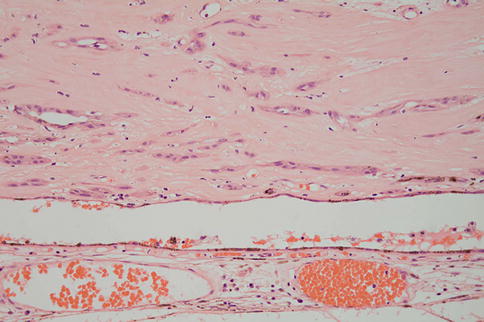
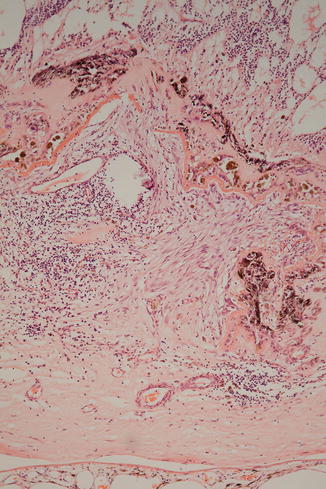
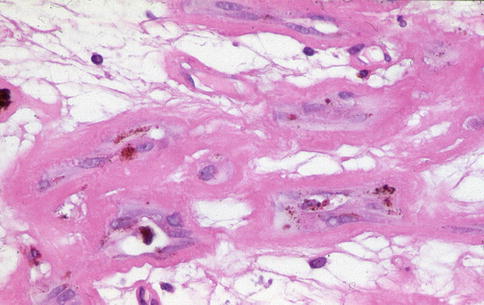
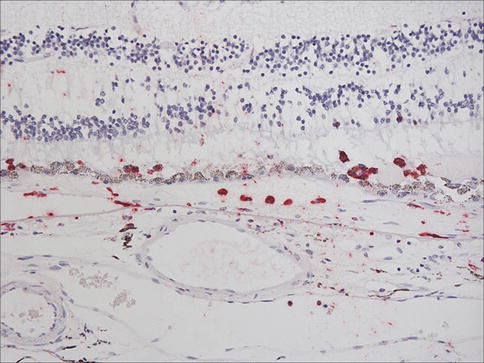
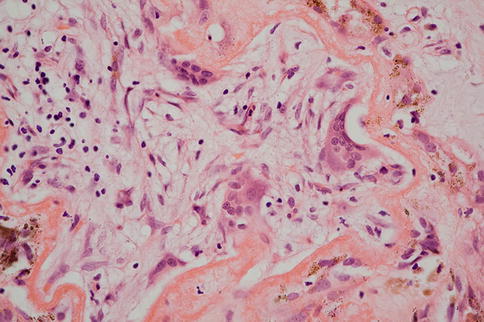
Fig. 9.24
Choroidal neovascular membrane between the RPE and Bruch’s membrane. Note there is also some subretinal fluid
Fig. 9.25
Islands of largely depigmented RPE within a relatively hypocellular, densely fibrotic neovascular membrane. In this instance the membrane lies internal to host RPE which is atrophic and has migrated under the membrane
Fig. 9.26
A large subretinal neovascular complex with one component between disorganized neural retina at the top and an irregular remnant of basal laminar deposit and another, occupying most of the field, between the basal laminar deposit and RPE and Bruch’s membrane and choroid at the bottom of the image
Fig. 9.27
Basal laminar deposit-like material being deposited by RPE that has migrated into a neovascular membrane
Fig. 9.28
Macrophages in association with a choroidal neovascular membrane (CD68 – red)
Fig. 9.29
Multinucleated giant cells associated with basal laminar deposit-like material in a choroidal neovascular membrane
If CNV membranes are excised surgically and submitted for examination (Fig. 9.30), it can be difficult to orient them accurately as the RPE can migrate over the undersurface of the membrane (Fig. 9.25). Indeed, drusen-like structures may form upside down if RPE migrates over the underside of the neural retinal (Fig. 9.31). If an opinion is to be given as to whether the membrane is subretinal or sub-RPE, it is helpful if either photoreceptor outer segments (or sometimes inter-photoreceptor “ghost” matrix (Fig. 9.24)) or Bruch’s membrane is present on one side or the other. It is also important to remember that combined membranes are not uncommon and that this may not be apparent unless step or serial sections are taken through the entire membrane.
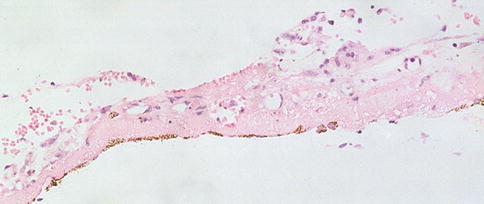
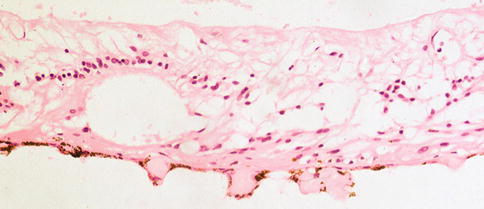
Fig. 9.30
An excised subretinal membrane with RPE that has possibly migrated, partially lining one aspect
Fig. 9.31
Drusen-like structures on the underside of gliotic retina, the RPE having migrated across the subretinal space
The retina over a membrane, which if fibrotic as is usually the case if they reach a histopathologist and is often known as a disciform scar, is predictably atrophic. Bruch’s membrane is remarkably resilient and is usually identifiable even in a profoundly disorganized posterior pole. A PAS stain may help. The choriocapillaris is often atrophied under a scar, and, indeed, choriocapillaris dropout may be an important early step in the pathogenesis of a scar and contribute to the hypoxic drive supporting new vessel formation.
In some instances, it is massive intraocular hemorrhage (Fig. 9.32) that precipitates enucleation, and the eye may be removed because it is not possible to ascertain whether or not there is a covert posterior pole neoplasm. Extensive organization of the hemorrhage can confound interpretation, but careful examination of multiple blocks and levels is usually sufficient. If there is major bleeding, particularly if the choroid is involved, the possibility of polypoidal choroidal vasculopathy should be considered. This condition, which is linked to but possibly separate from AMD, is more common in Asians although currently likely to be underdiagnosed in Western populations. The presence of large dilated polypoidal vessels in the choroid is the key to histopathological diagnosis.
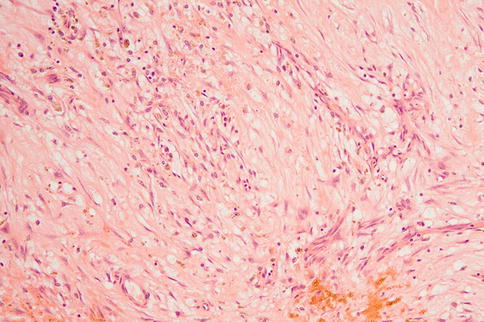
Fig. 9.32
Organized intraocular hemorrhage
Geographic atrophy is characterized by combined loss of RPE, photoreceptors, and choriocapillaris with in extreme cases downstream atrophy of inner retina (Fig. 9.33). There is a perhaps surprising variability in the concordance of the photoreceptor, RPE, and choriocapillaris degenerations, and it is certainly a challenge to identify which element degenerates first. It may be better to consider geographic atrophy a “system” disorder involving all the components of the retina–RPE–Bruch’s–choroid complex. As with other epithelia, the RPE strives to maintain integrity over the surface of Bruch’s membrane in the face of even marked cell loss. There can be marked attenuation with loss of pigment including lipofuscin. It is therefore possible that RPE remains even when the autofluorescence signal on fundus imaging is lost.
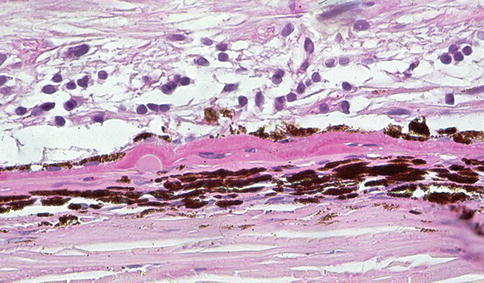
Fig. 9.33
Marked retinal atrophy (the nerve fiber layer is at the top of the figure although the inner limiting membrane is not included). There is patchy loss of RPE and a small druse as well as basal laminar/basal linear deposit that contribute to the separation of RPE from Bruch’s membrane. The choroid is atrophic
The term reticular pseudodrusen relates to a clinical appearance at the posterior pole characterized by ribbonlike areas of alternating light and dark and scattered, small, ringlike lesions, often with a central spot. These are best seen using near-infrared reflectance imaging. It has been proposed that these correspond to subretinal drusen-like deposits that are seen on OCT imaging, but the interpretation of these images, which are computer reconstructions, is a little unclear and although subretinal debris has been seen histopathologically, definitive clinicopathological correlation is not yet available.
Special stains, including immunohistochemistry, are of limited value in the assessment of these cases. A periodic acid–Schiff stain can highlight some of the deposits and Bruch’s membrane, and a vascular marker can make it easier to assess the loss of choriocapillaris (which is sometimes masked by the presence of “holes” in the choroidal stroma that remain after the endothelium has died). A pan cytokeratin can assist in identifying migrated, depigmented RPE. There is sometimes ambiguity of staining with CD68 as RPE cells, particularly in pathological states, can express this marker as well as macrophages.
Differential Diagnosis
One main differential diagnosis of early AMD is of “normal” aging. If there are scattered small hard drusen without CNV or GA in an elderly patient, the findings are best considered those of aging alone. Large macular soft drusen with or without extensive sub-RPE deposits are indicative of AMD.
The differential diagnosis of advanced AMD is most often an issue in younger patients. Here it is important to consider rare inherited disorders such as Sorsby’s fundus dystrophy, Malattia leventinese, and late-onset retinal degeneration (L-ORD). These conditions lead to thick, extensive sub-RPE deposits and CNV. Sorsby’s fundus dystrophy also leads to deposition of abnormal extracellular material over the epithelium of the pars plicata of the ciliary body, not something seen in AMD. A variety of other rare conditions can lead to diffuse deposit formation, but these are hardly ever an issue. There are very many causes of choroidal neovascularization, and before assuming that it is due to AMD, it is important to consider these. Pathological myopia leads to an enlarged eye, breaks in Bruch’s membrane, and CNV. Almost any inflammatory process, infectious, or otherwise, at the back of the eye can result in CNV as can trauma, laser photocoagulation, radiation retinopathy and other degenerative changes in Bruch’s membrane such as angioid streaks arising in pseudoxanthoma elasticum. What is a challenging issue is how to classify CNV in an elderly patient without obvious other histopathological or clinical features of AMD. This may be designated idiopathic, but this may turn out to be a genuine subtype of AMD as it becomes clearer what the pathogenesis of the condition truly is.
Genetics
The genetics of AMD is now understood in great detail, remarkably so for a complex age-related disorder. A significant component of the risk of developing disease rests with polymorphisms in the complement factor H (CFH) locus on chromosome 1 and with polymorphisms at the ARMS2/HTRA1 locus on chromosome 10. Other genes that contribute to risk in some individuals include other genes with a role in complement activation, other aspects of innate immunity or inflammation, genes associated with high-density lipoprotein metabolism, and several rare variants. Furthermore, copy number variation, epigenetic factors, and mitochondrial genetics may be important.
Prognosis and Predictive Factors
The main environmental risk factor for AMD is smoking, and there is evidence to suggest that a healthy diet is protective, particularly for those with a high-risk genotype, whereas elements of a “Western” diet associated with a poor cardiovascular risk also are adverse risk factors for AMD.
9.6.2 Retinal Detachment
Definition
Retinal detachment arises in a number of circumstances that all lead to separation of the neural retina from the retinal pigment epithelium (RPE). Rhegmatogenous retinal detachment arises when the retina is disrupted, relatively rarely as a result of trauma to the eye and more often due to the development of tractional forces between the retina and vitreous. Fluid from the vitreous cavity thereby gains access to the subretinal space, and RPE may access the outer retinal surface. In non-rhegmatogenous retinal detachment, the retina remains intact. A common cause of the latter is when exudate accumulates between the retina and RPE. For many pathologists, this is most often seen when examining eyes with choroidal melanomas. In central serous retinopathy, apparently spontaneous accumulation of subretinal fluid is seen, often in young patients. Tractional retinal detachment develops where preretinal membranes, with or without a vascular component, display typical contraction associated with a wound healing process.
Epidemiology
Relatively little is known of the epidemiology of retinal detachment although a recent meta-analysis gave an incidence of approximately 6–18 per 100,000 population [33].
Etiology
As discussed above, trauma is an important cause of retinal detachment and, whether associated with trauma or other processes at the vitreoretinal interface such as diabetic neovascularization, in many instances the retina is pulled away from the RPE by traction, essentially of contracting vitreous collagen on the inner limiting membrane of the retina. Retinal detachment may be preceded by vitreous detachment. Certain disorders of the peripheral retina such as lattice degeneration and holes and tears that form without obvious cause as well as macular hole (see below) all predispose to retinal detachment [34]. High myopia with enlargement of the eye is another risk factor. Accumulation of material under the retina is a further cause of detachment. Commonly, exudative detachment is seen adjacent to a choroidal melanoma because of the leakage of fluid from the tumor. A similar process operates over many choroidal neovascular membranes.
Clinical Features
Depending on the location and cause, patients may experience the sudden appearance of “floaters” or flashing lights. They may witness a “shadow” appearing across their vision.
Macroscopy
The outside of the eye should be examined for evidence of encircling bands, plombs, or other forms of surgical intervention. When the eye is opened, there is sometimes an outpouring of silicone oil that has been used surgically to keep the retina flat following a vitrectomy. The intraocular macroscopic findings may be dominated by other pathology, for instance, an underlying disciform scar, proliferative vitreoretinopathy, or choroidal tumor. In other instances, it may be difficult to distinguish a real detachment from an artifactual one, or a very careful examination of the peripheral retina for an area of lattice degeneration is required.
Histopathology
The retina often rapidly becomes edematous (Fig. 9.34), and with time, loss of outer segments gives way to outer retinal atrophy. Gliosis may be marked, and Müller cells may extend and/or migrate into the subretinal space (Fig. 9.35) preventing reattachment. Similarly, they can move onto the anterior retinal surface (Fig. 9.36), often passing through the inner limiting membrane just anterior to retinal vessels. Particularly if there is a retinal break, the preretinal proliferation may include migrating RPE cells which can then, with the glial cells, generate a complex membrane capable of contraction and exacerbating the clinical situation.
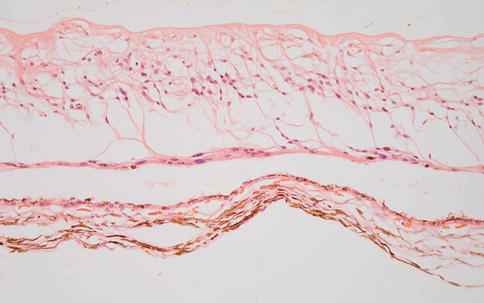
Fig. 9.34
Multiple layers of retina folded across each other in an eye removed shortly after traumatic retinal detachment. The outer plexiform layer especially (middle of the figure) is edematous (PAS)
Fig. 9.35
Glial cells have migrated along the underside of the retina to form a scar. There is artifactual separation between this and the subjacent RPE
Fig. 9.36
A thin glial membrane is seen bridging two parts of the inner limiting membrane. Note the presence of edema with proteinaceous fluid in microcysts in the inner nuclear layer and outer plexiform layer
In long-standing retinal detachment, the retinal gliosis becomes very marked, and the normal laminar appearance of the retinal can be lost (Fig. 9.37). There may be some intraretinal mineralization, and RPE may migrate into the retina and as in other retinal degenerations such as retinitis pigmentosa (see above) aggregate around retinal blood vessels.
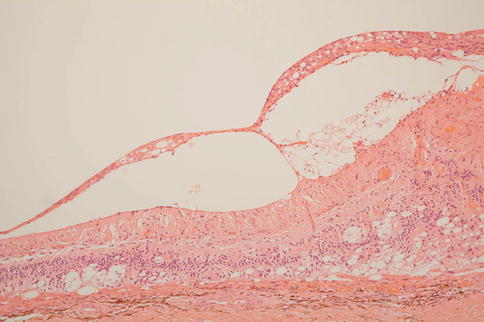
Fig. 9.37
Long-standing retinal detachment with disorganization and gliosis of the retina. Note the presence of an epiretinal membrane that is apparently causing retinal traction. The retina is, however, attached
If the detachment is large enough, it can lead to a so-called funnel-shaped detachment with retina gathered up behind the lens (Fig. 9.38), if present, and a tube of retina extends posteriorly to the optic nerve head.
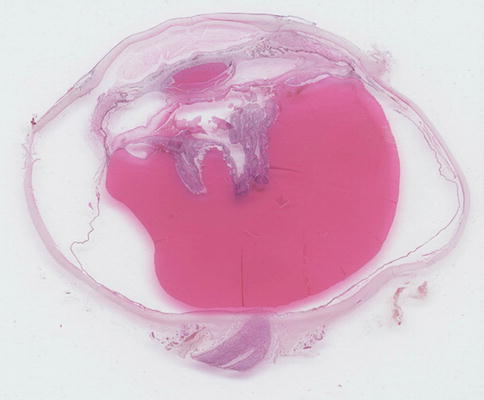
Fig. 9.38
A complete section of a globe with extensive retinal detachment, the subretinal space filled with proteinaceous fluid, and the retina itself gathered up behind the lens
The composition of the material in the subretinal space can vary depending on the cause of the detachment. Adjacent to a choroidal melanoma, there is usually rather homogeneous protein-rich fluid (Fig. 9.39). In Coats’ disease there is typically an abundance of foamy macrophages and cholesterol clefts. The RPE may round up or migrate. In long-standing detachment, it is often the case that drusen-like sub-RPE deposits are present. These drusen-like structures are often arranged in clusters (Fig. 9.40).
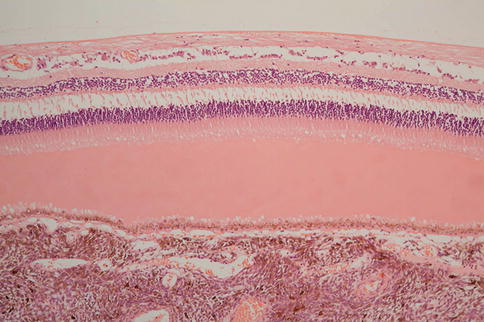
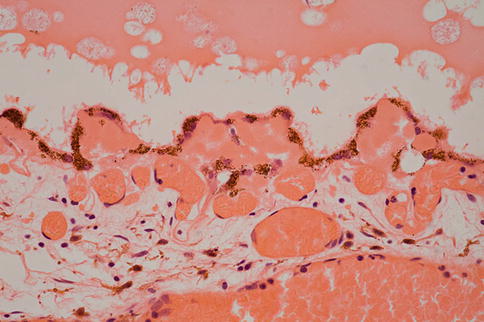
Fig. 9.39
Protein-rich exudate separating retina from RPE over a choroidal melanoma. The layer of fluid is relatively thin, and there remains preservation of some photoreceptors and their outer segments
Fig. 9.40
Drusen-like deposits under the RPE in a case of long-standing retinal detachment
There may be evidence of underlying causal pathology. For instance, there may be a healed scleral wound indicative of previous trauma or abnormal preretinal vessels indicative of proliferative diabetic retinopathy or retinopathy of prematurity.
There may also be evidence of therapeutic intervention ranging from subtle pars plana scars at the site of vitrectomy through to encircling bands (Figs. 9.41 and 9.42) and silicone oil. The latter can reside within the eye and cause relatively little damage, or it can provoke a widespread, macrophage-rich inflammatory cell infiltrate. Oil can extend as far posteriorly as the optic nerve head and anterior to the drainage apparatus (Figs. 9.43 and 9.44) may even exit anteriorly through trabeculectomy wounds into the subconjunctival space. It is sometimes difficult to distinguish intraretinal degenerative cystic change from spaces due to oil. A useful pointer to the latter is the presence of RPE granules, which impart a “dirty” quality to the vacuoles (Figs. 9.45 and 9.46). It is possible that the RPE granules are in part responsible for emulsifying the oil.
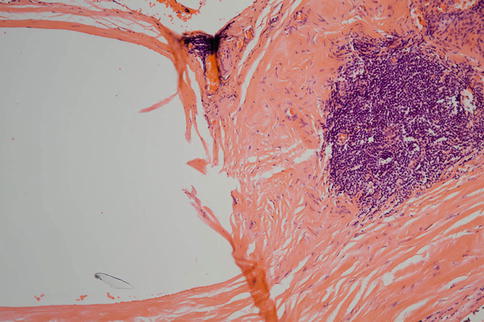
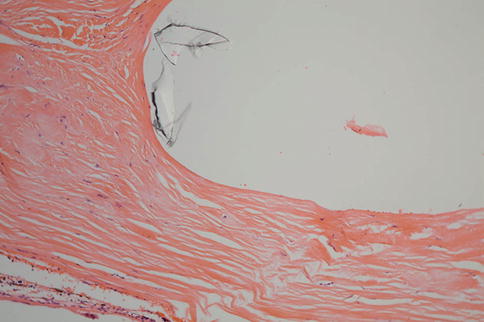
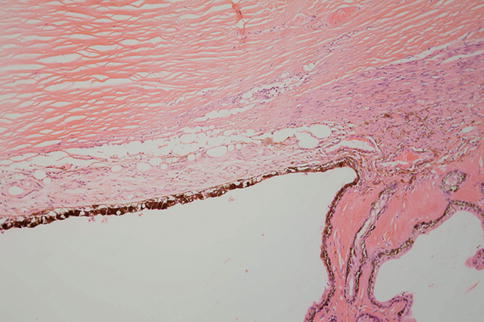
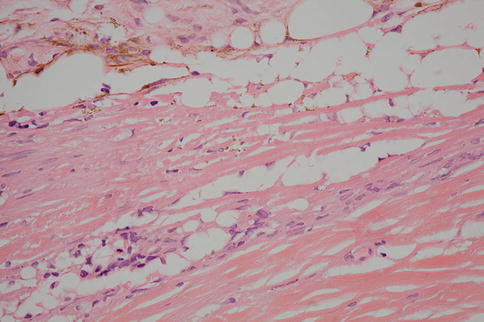
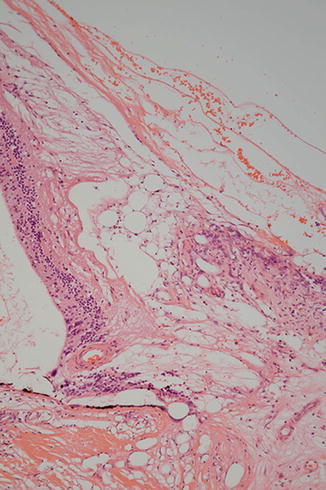
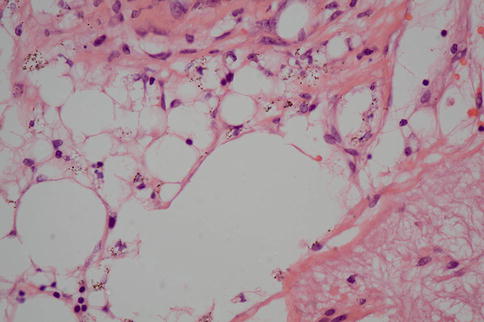
Fig. 9.41
The cavity on the left of the figure was occupied by an encircling band from a retinal detachment procedure. On the right, a focus of chronic inflammatory cell infiltrate is present in the scarred episcleral tissue
Fig. 9.42
An encircling band similar to Fig. 9.26 with some thinning of the sclera and some of the material from the band apparent in the space in the upper right portion of the figure
Fig. 9.43
An occluded anterior chamber and chamber angle due to low-grade inflammation associated with silicone oil droplets. Some oil is probably also present in the drainage system
Fig. 9.44
High-power image of silicone oil, evident as round optically empty spaces, present in the anterior chamber angle (top), trabecular meshwork, and proximal drainage apparatus (bottom left)
Fig. 9.45
Tractional retinal detachment close to the optic nerve head (bottom right) with numerous droplets of silicone oil
Fig. 9.46
High-power image of silicone oil droplets. Note how the admixture of RPE pigment granules gives a “dirty” appearance to the droplets. This can be helpful in distinguishing oil from microcystic degeneration in the retina
Differential Diagnosis
The retina often separates artifactually from the RPE following removal or during processing. This is easily distinguished from true detachment because there is no material visible in the subretinal space; usually the photoreceptor outer segments are present (unless there is some other disease), and the outer segment tips are lightly decorated with elongated pigment granules from the apical processes of the RPE (Fig. 9.47). Distinguishing between different causes of retinal detachment often requires clinicopathological correlation, and certainly, in end-stage eyes, it can be very difficult to ascertain what the primary pathology was.
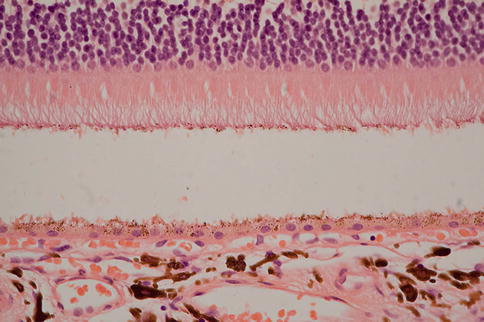
Fig. 9.47
Artifactual retinal detachment. Note the well-preserved outer segments and how they are decorated with RPE pigment from disrupted RPE apical microvilli
Genetics
A variety of conditions with a genetic basis are associated with retinal detachment. These include Stickler’s syndrome (COL2A1, COL11A1, COL11A2, and COL9A1), Coats’ disease (in which there may be mutations in norrin), and familial exudative vitreoretinopathy (frizzled–4). For other conditions such as retinopathy of prematurity and lattice degeneration, there are genetic risk factors. A recent GWAS study has identified high-risk loci for rhegmatogenous detachment [35].
9.6.3 Myopia
Definition
Clinically, myopia is short-sightedness where the image forms only in a plane anterior to the photoreceptors. It may be classified into two broad categories. One, axial myopia, arises because the eye is abnormally long. The other, refractive myopia, arises because either the refractive index of the optical media is altered or because of abnormalities of curvature of one or more optical surfaces in the eye, typically the cornea. This section will describe the changes in the eye in the former type. Significant pathology only tends to arise where there is so-called high myopia where the refractive abnormality is greater than 6.0 diopters. It is in this group that changes that a pathologist can identify tend to become apparent.
Synonyms
Myopia, short-sightedness, and near-sightedness are all the same. The terms pathological or degenerative myopia are sometimes used as a synonym for high myopia, but they are perhaps better reserved for instances where pathological changes beyond increased axial length are apparent.
Epidemiology
Myopia is very common, but pathological myopia probably develops in about 2 % of myopic patients overall. However, the prevalence appears to be much higher in Asian populations. In East Asia it affects 80–90 % of school leavers with 10–20 % of those completing secondary education suffering from high myopia [36].
Etiology
The etiology of myopia is complex and not fully understood. There is no doubt that genetics are important and there do appear to be important racial differences, but environmental factors appear to be of dominant significance. Urbanization with relative lack of time spent outdoors seems to be of major importance. It may be that lack of green cues is particular critical.
Clinical Features
The defining clinical feature is abnormal refraction, and increased axial length can be determined by ultrasound. Fundus examination may disclose posterior vitreous detachment, a crescent of apparent depigmentation adjacent to the optic nerve head, retinal detachment due to lattice degeneration or retinal holes, myopic degeneration with punched out areas of retinal, RPE and choroidal degeneration or lacquer cracks which represent breaks in Bruch’s membrane. These breaks may give rise to choroidal neovascularization, and associated subretinal fibrosis with pigment migration is known as a Fuchs’ spot.
Macroscopy
The normal eye has an axial length of 24–5 mm. If it is larger than this, the possibility of myopia should be considered. In severely myopic eyes, the axial length may increase to 29 mm or more. Generally the eye becomes elongated, so horizontal and vertical diameters are usually normal. The expansion of the sclera is often most marked posteriorly, and there may be a distinct bulging of the sclera known as a staphyloma. The stretching leads to scleral thinning which may make the eye difficult to open. Irregular expansion of the eye is more likely to be associated with pathological myopic changes, but such irregularities can be harder to assess in the enucleated eye because of the loss of intraocular pressure. Intraocular examination may appear to be relatively normal or any of the clinical features listed above may be apparent. Peripheral cobblestone degeneration may be more common in myopia, but it is present in a significant proportion of eyes over 20 years of age without myopia.
Histopathology
The histopathological findings in a myopic eye are often complex and include the complications of myopia and any subsequent therapy. For instance, retinal detachment may then be complicated by proliferative vitreoretinopathy. The features to be focused on in this section are those arising primarily from the myopia itself.
The histopathology of myopia has been described thoroughly by Grossniklaus and Green [37]. A common feature, seen in over a third of their cases, is the temporal displacement of the optic nerve head. This leads to choroid overriding the nasal nerve head and a shift of choroid, retinal pigment epithelium, and neural retina away from the temporal margin of the optic nerve head. Where the sclera is stretched, there is thinning of the neural retina and choroid (Fig. 9.48a), and there may be secondary holes or schisis cavities in the retina that may predispose to retinal detachment (see above). Areas of atrophy may be quite striking with extreme thinning of the retina and choroid with loss of retinal pigment epithelium. Breaks in Bruch’s membrane (lacquer cracks) may be found rarely, and choroidal neovascularization where present is more apparent.
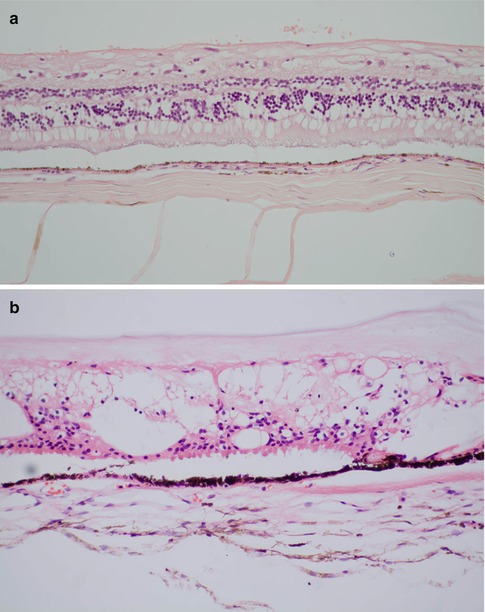
Fig. 9.48
(a) Thinning of retina and choroid in a myopic eye. (b) Peripheral microcystoid degeneration of the retina
Differential Diagnosis
Each of the components of the myopic eye has its own differential although in general once the different elements have been put together there is no great diagnostic challenge. The optic nerve head changes need to be distinguished from glaucomatous change. Although the peripapillary pallor seen clinically may be mistaken for peripapillary atrophy when viewed en face, it is seen to be displacement and not atrophy histologically. Any areas of atrophy and choroidal neovascularization must be distinguished from age-related macular degeneration and posterior staphylomas from other causes such as inflammation or buphthalmos.
Genetics
GWAS studies [38] have identified a significant number of genes related to extracellular matrix modeling, neural transmission, retinoic acid metabolism, and eye development that explain a modest degree of the variation in expression of myopia. Further studies, particularly with exome sequencing, are likely to generate further insights, but it seems probable the detailed pathogenesis of myopia will be complex.
9.6.4 Retinal Holes, Tears, Schisis, and Related Degenerations
Definitions and Clinical Features
The retina is vulnerable to a wide range of acquired structural degenerations or defects, some of which may predispose to retinal detachment. Others are of no particular clinical importance.
Retinal holes may be incidental defects that may or may not be associated with other degenerative abnormalities of the retina or adjacent vitreous. They may be associated with rhegmatogenous retinal detachment. Another clinically important form of retinal hole is the macular hole, which forms centered on the fovea. Lattice retinal degeneration is usually found in the peripheral retina. The abnormal area of retina may contain foci of heaped up retinal pigment epithelium and pale hyalinized blood vessels.
Cobblestone or paving stone degeneration is commonly seen just behind the ora serrata and consists of irregular, small pale patches. Also common and just posterior to the ora serrata is peripheral microcystoid degeneration, which has a fine honeycomb appearance. This is generally without consequence although it can lead to retinoschisis, splitting of the retina in a plane parallel to the inner limiting membrane, and retinal detachment.
Histopathology
The edges of retinal holes are generally rounded and can be distinguished from artifactual tears. Macular holes evolve through a series of stages and may be associated with a tiny operculum just anterior to the hole, and as with other structural retinal defects, there is often apparent vitreous traction. Vitrectomy, sometime with stripping of the inner limiting membrane, may cause the hole to close, supporting the notion that vitreous traction is important in pathogenesis. In peripheral microcystoid degeneration, small cysts appear first in the outer plexiform layer and then more anteriorly (Fig. 9.48b). In cobblestone degeneration, there is full-thickness atrophy of retina, retinal pigment epithelium and choroid.
9.7 Retinal Vascular Disease
Retinal vascular disease covers a wide range of inherited and acquired conditions, some of which have been included elsewhere. This section will focus on vascular changes associated with diabetes mellitus and hypertension as well as retinal artery and retinal vein occlusions.
9.7.1 Diabetic Retinopathy
Definition
Diabetic retinopathy can be considered in three phases. Late-stage proliferative diabetic retinopathy is characterized by the presence of newly formed blood vessels, typically anterior to the retina. Pre-proliferative or background retinopathy has no such vascular proliferation, but small hemorrhages, exudates, and cotton wool spots are evident. It is useful to also recognize an even earlier phase where no obvious clinical or histopathological abnormalities are present, yet it is clear that there are functional and probably very subtle structural changes. In this section the focus will be on pre-proliferative and proliferative diabetic retinopathy. A significant cause of visual impairment is the development of diabetic macular edema, and a predominantly ischemic form of the disease with progressive enlargement of the foveal avascular zone is also recognized.
Epidemiology
It is well established that diabetes, especially type II diabetes, is increasing in prevalence in industrial societies at an alarming rate over much of the world, and diabetic retinopathy is set to follow [39].
Etiology
Elevated blood sugar levels, other metabolic factors and, where present, coincident hypertension all contribute to the pathology seen in the eye.
Clinical Features
Patients with early disease are likely to be visually asymptomatic, and it is for this reason that screening programs for diabetic retinopathy can be so effective. With advancing disease, visual acuity can drop progressively, or if there is significant vitreous hemorrhage, it can drop precipitously.
Macroscopy
In an enucleated eye, it can be difficult to see focal pathology, but cotton wool spots and small retinal hemorrhages may be evident, but such eyes are very rarely received. More often, vitreous hemorrhage has provoked enucleation and this is readily identified or the eye has become phthisical and end stage where macroscopic examination confirms disorganized intraocular contents but generally offers little if any insight into the underlying pathology. Evisceration specimens are equally hard to access.
Histopathology
The histopathology of the diabetic eye is often complex, particular in advanced disease where there has been therapeutic intervention. Eyes with nonproliferative disease are rarely submitted for examination but may be received incidentally because of, for instance, an exenteration for anterior malignancy. Anterior changes can include rubeosis iridis (Fig. 9.49), lacy vacuolization of the iris pigment epithelium (due to glycogen accumulation), cataract, and thickening of the basement membrane of the ciliary epithelium (Fig. 9.50).
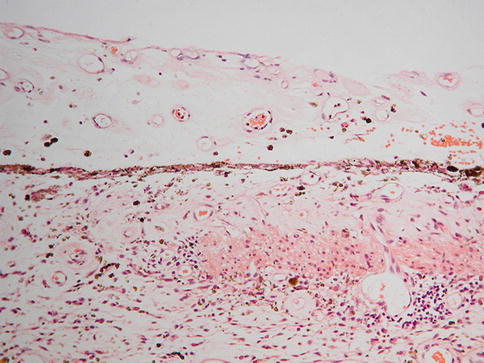
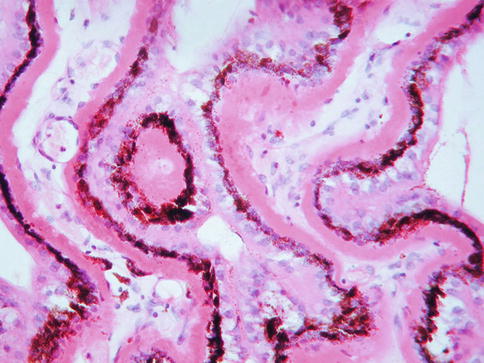
Fig. 9.49
Rubeosis iridis with a pronounced neovascular layer along the anterior surface of the iris (top). The iris stroma is somewhat edematous, and there is a patchy chronic inflammatory cell infiltrate
Fig. 9.50
Thickening of the basement membrane of the pigmented epithelium of the ciliary body in the eye of a diabetic patient (PAS)
In very early disease, the retina may appear normal by conventional microscopy. If cotton wool spots or other focal pathology are identified macroscopically, they are more readily sampled for microscopic examination than if “random” levels are taken.
Cotton wool spots are essentially focal areas of ischemia in the nerve fiber layer. Axonal transport is impaired, and as a result, cytoplasmic organelles and other material accumulate at the site of hypoxia resulting in swelling of nerve fibers, which are known as “cytoid bodies” (Fig. 9.51). Cotton wool spots disappear after a few weeks leaving a barely discernible glial scar in the nerve fiber layer. Intraretinal hemorrhages again are straightforward to diagnose when present.
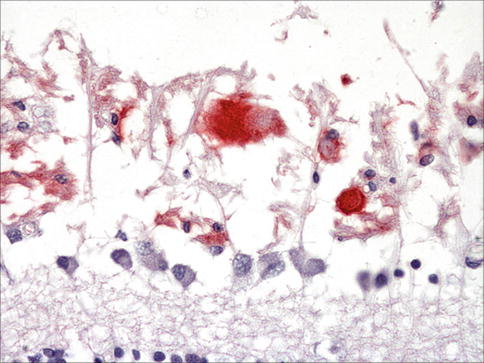
Fig. 9.51
The nerve fiber layer from a patient with SLE and extensive ischemic change within the retina. The swollen axons are demonstrated with immunohistochemistry for a mitochondrial protein, which highlights the so-called cytoid bodies. Note a row of ganglion cells and the inner plexiform layer at the inferior aspect of the image
It is often stated that thickening of microvascular basement membrane is an early and significant feature of diabetic retinopathy [40]. This is, however, surprisingly difficult to demonstrate in routine preparations, even with PAS staining although when present can be quite striking (Fig. 9.52). What is not infrequently seen is thickening of the vessel wall in a way that is similar if not identical to that seen in hypertension, and, of course, hypertensive disease is quite likely to coexist. Also difficult to demonstrate is the presence of pericyte degeneration, another early and probably significant change in diabetes. Interestingly, the retinal microvessels in health have an unusually high ratio of pericytes to endothelial cells, and this may be important in the local control of retinal blood flow.
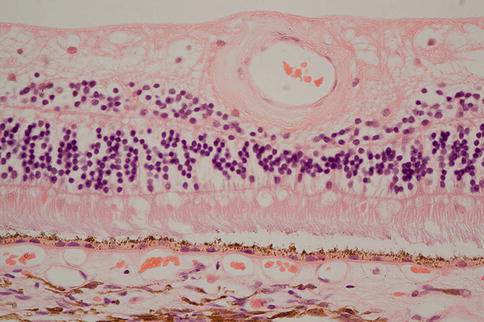
Fig. 9.52




Thickening of the wall of a retinal arteriole in a patient with diabetes mellitus
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
