9 ![]()
Definition of the Problem
What Is Pigmentary Glaucoma?
Pigmentary glaucoma is a form of secondary open-angle glaucoma, which is characterized by the presence of radial midperipheral iris transillumination defects, pigment deposition on the corneal endothelium (Krukenberg’s spindle), and dense pigmentation of the trabecular meshwork. When these slit-lamp findings are present without elevated intraocular pressure (IOP), the condition is known as pigment dispersion syndrome.
When Was Pigmentary Glaucoma First Described?
Krukenberg1 initially described the vertical, central pigment deposition on the corneal endothelium, which now bears his name, in 1899. Later authors such as Von Hippel2 and Jess3 hypothesized that pigment dispersion may contribute to elevated IOP in glaucoma patients. The term pigmentary glaucoma was first used in the 1940s when Sugar and Barbour4 described two myopic glaucoma patients with excessive pigmentation of the trabecular meshwork. Later authors identified the source of this pigment as the neuroepithelium along the posterior surface of the iris, accounting for the characteristic transillumination defects.5,6
Epidemiology and Importance
Who Is at Risk for Pigmentary Glaucoma?
Pigmentary glaucoma typically occurs in young adults. Although pigment dispersion syndrome is as common or more common in women than men, pigmentary glaucoma is typically found in men.7,8 Moreover, the development of glaucoma is usually earlier in men, with an average age of onset of 34 to 46 years, compared to women, who have an average age of onset of 46 to 53 years.7,8 The most common refractive error seen in pigmentary glaucoma is mild to moderate myopia.8 Berger and associates9 reported that higher degrees of myopia may lead to an earlier age of onset of glaucomatous optic nerve damage in patients with pigmentary glaucoma.
Although pigmentary glaucoma is characteristically found in whites, it has also been reported in a black albino patient10 and a mulatto family.8 In their large review of the risk factors for the development and severity of glaucoma in the pigment dispersion syndrome, Farrar et al11 examined the medical records of 93 patients with pigmentary glaucoma and 18 patients with pigment dispersion syndrome. They identified male gender, black race, severe myopia, and presence of Krukenberg’s spindle as possible risk factors for the presence of secondary glaucoma. Men made up two-thirds of this population and developed glaucoma at an earlier age requiring more extensive therapy. Although only four black patients were found in this study, they displayed a particularly aggressive form of the disease and all required surgical intervention.
Although pigmentary glaucoma typically occurs in young adults, it may also be found in older patients as well. Because the clinical manifestations of the disorder may diminish with time, older patients may present with normalized IOP and regressed pigmentary changes, but showing optic nerve damage, leading to an erroneous diagnosis of normal tension glaucoma.12
What Are the Genetic Characteristics of Patients?
Our understanding of the genetics of pigmentary glaucoma continues to unfold. Although many cases of pigmentary glaucoma and pigmentary dispersion syndrome are sporadic, an early report by Stankovic13 suggested an autosomal recessive pattern of inheritance for the disorder. A more recent article by Olander and co-workers14 supports transmission by either an autosomal dominant or recessive pattern. Support for a genetic basis for the disease comes from its observation in siblings and twins, and the fact that there is an increased incidence of open-angle glaucoma in the families of patients with pigment dispersion syndrome. Pigment dispersion patients also show an increased incidence of steroid responsiveness, indicating a possible underlying predisposition for open-angle glaucoma. Andersen and associates15 studied 54 members of four families with pigment dispersion syndrome and pigmentary glaucoma. Twenty-eight patients showed clinical evidence of the pigment dispersion syndrome, of whom 14 required medical or surgical treatment. These authors found the pigment dispersion syndrome to be inherited as an autosomal dominant trait in these four families, and they linked the gene responsible for the syndrome to the telomeric end of the long arm of chromosome 7q35-q36. Although still very early in development, the isolation and characterization of the gene responsible for pigment dispersion syndrome may someday help us better understand the pathophysiology of the disorder and lead to new methods of diagnosis and treatment.
How Common Are the Pigment Dispersion Syndrome and Pigmentary Glaucoma?
The pigment dispersion syndrome has been found to be much more common than was originally reported.4 Ritch and associates16 performed two large population screenings, which included slit-lamp examination, and detected pigment dispersion syndrome in 18 of 934 individuals for an overall prevalence of 1.93%, and 2.45% of white patients. The authors noted that many of these patients displayed a very mild form of the syndrome and concluded that many similar patients probably go undetected in their lifetime. Likewise, pigmentary glaucoma is not uncommon as well, and may account for up to 1.5% of all glaucomas worldwide.17
The percentage of patients with pigment dispersion syndrome who develop pigmentary glaucoma is unclear. An early report by Wilensky and co-workers18 found that only two of 43 patients who presented with Krukenberg’s spindle and normal visual fields developed visual field loss after 5.8 years. More recent longitudinal studies, however, have suggested that up to 50% of patients with pigment dispersion may eventually develop pigmentary glaucoma.11,19,20 Migliazzo and associates19 reported a retrospective, longitudinal review of 129 eyes in 65 patients with pigment dispersion syndrome. Visual acuity, IOP, visual field loss, pigment grade, Krukenberg’s spindle, iris transillumination defects, medications, and surgeries were analyzed for each patient at 5-year intervals for an average follow-up period of 17 years. Optic disc and visual field changes developed in 35% of patients with pigment dispersion and ocular hypertension during the study period. Richter and co-workers20 performed a prospective study of the natural history of pigment dispersion syndrome and pigmentary glaucoma in 55 patients for an average follow-up of 27 months. They observed active pigment dispersion, defined by increase in iris transillumination or corneal pigmentation or by the appearance of pigment granules on the surface of the lens, in 45 eyes of 31 patients and was associated with worsening of glaucoma in 32 eyes. They found no differences in the frequency of active dispersion of pigment and worsening of glaucoma with respect to age, even in those patients older than 65 years. The authors concluded that progression of pigmentary glaucoma is correlated with active pigment dispersion and may continue to occur in older patients.
Other risk factors for the development of glaucoma in pigment dispersion patients include male gender, severe myopia, and presence of Krukenberg’s spindles11 (Table 9–1). Once present, pigmentary glaucoma may be more difficult to control than primary open-angle glaucoma (POAG). In a large series, Scheie and Cameron8 found surgical intervention was required in 23.5% of pigmentary glaucoma patients compared with 14.6% of patients with POAG.
Male gender |
White race |
Young age (3d to 4th decade of life) |
Mild to moderate myopia |
Diagnosis and Differential Diagnosis
What Are the Clinical Features of Pigmentary Glaucoma?
Pigmentary glaucoma and the pigment dispersion syndrome are characterized by a central vertical deposition of pigment along the corneal endothelium. Aqueous convection currents from the nasal and temporal halves of the anterior chamber result in the typical distribution pattern, with more pigment collecting inferiorly than superiorly. The endothelial cells phagocytose the pigment and apparently maintain their normal function, as normal endothelial cell counts and corneal thicknesses have been reported in pigment dispersion syndrome.21 Pigment may also accumulate along the anterior iris surface, leading to the development of heterochromia in cases of asymmetric pigment dispersion.22 Midperipheral iris transillumination defects, seen by scleral transillumination or retroillumination at the slit lamp, are characteristic findings in pigmentary glaucoma. The defects, which correspond in location to the underlying anterior lens zonules, may vary in severity from a few spoke-like changes to 360-degree involvement of the iris.
Pigment may be deposited on the posterior lens capsule and tends to collect in a ring corresponding to the location of the attachment of the zonules. This “Scheie’s line” is considered to be pathognomonic for pigment dispersion syndrome.23 Individual pigment particles may also be seen on the anterior lens capsule and anterior zonules.20
Pigment also collects in the angle structures, especially within the trabecular meshwork. This is seen gonioscopically as a heavy, dark discoloration over its entire circumference. A pigmented Schwalbe’s line, or Sampaolesis’ line, is also typically seen. Several authors have described an increased number of iris processes in patients with pigmentary glaucoma.24,25 The ciliary band is typically seen easily on gonioscopy as expected in a large, myopic eye. Clinical features of pigment dispersion syndrome and pigmentary glaucoma are listed in Table 9–2.
How Do Exercise and Pupillary Dilation Affect Pigmentary Glaucoma Patients?
Patients with pigment dispersion syndrome or pigmentary glaucoma may shed excess pigment following exercise26 or pupillary dilation,27 with a resultant increase in IOP. This induced pigment release and IOP elevation can be blocked by pilocarpine28 and partially blocked by laser iridotomy.29 Shenker and associates26 describe a 32-year-old white man with pigment dispersion syndrome who developed a significant rise in IOP and anterior chamber pigment release shortly after playing basketball. The authors concluded that active pigment dispersion caused by exercise led to an acute rise in IOP.
Pigment dispersion syndrome |
Krukenberg’s spindle |
Midperipheral iris transillumination defects |
Excess pigmentation of the angle |
Pigmentary glaucoma |
All of the above, plus: |
Elevated intraocular pressure |
Glaucomatous optic disc atrophy |
Characteristic visual field changes |
Exercise, however, does not always lead to an increase in IOP in pigmentary glaucoma patients. Smith and co-workers30 measured IOP in 10 pigmentary glaucoma patients following a 25-minute exercise protocol. Applanation tonometry measurements during the first 2 hours after exercise showed no statistically significant rise in IOP in any of the patients. Other authors have reported a decrease in IOP in pigmentary glaucoma patients following exercise.31 The mechanism for decreased IOP following exercise is not clear, although some authors have suggested increased plasma osmolarity32 or increased facility of outflow33 as possible mechanisms. Although exercise may not elevate IOP in all pigmentary glaucoma patients, those who complain of blurring vision or halos around lights following vigorous activity should be checked to see what, if any, IOP response they may have.
What Is the Pathophysiology of Pigment Dispersion Syndrome?
Initially, loss of iris pigment was thought to be congenital in origin, resulting from either the maldevelopment of the pigment layer with resultant atrophy34 or cellular hyperplasia of the muscular portion of the iris neuroepithelium.35 This hereditary developmental concept has been supported by the association of pigmentary glaucoma with abnormalities of the retinal pigment epithelium and peripheral retina.36,37 In 1979, Campbell38 described a new mechanism for pigment dispersion after noting the high prevalence of posterior iris bowing in pigmentary glaucoma patients. He proposed that friction between the posterior iris surface and underlying lens zonules, which occurs during normal pupillary movement, damages the epithelial cells and results in pigment release. This theory was later confirmed by Kampik and associates39 using scanning and transmission electron microscopy.
A reverse pupillary block mechanism in which the iris acts as a “flap valve” to maintain a pressure gradient between the anterior and posterior chambers was described by Karickhoff.40 In this hypothesis, the iris is pushed backward against the anterior lens zonules when a higher pressure exists in the anterior as compared to the posterior chamber of the eye. The reverse pupillary block mechanism has been well demonstrated by ultrasound biomicroscopy41 and can be relieved by laser iridotomy,29 with resultant iris flattening and loss of iridolenticular contact.
Several theories for the development of reverse pupillary block have been proposed. Campbell42 and Liebmann et al43 have shown that cessation of blinking in patients with concave irides results in iris flattening. This led them to propose that normal blinking may produce transient vector forces within the eye, causing aqueous to flow from the posterior to the anterior chamber. The iris then acts as a flap valve as described by Karickhoff40 to maintain a pressure gradient between the two chambers. The higher pressure in the anterior chamber then pushes the iris posteriorly, creating a reverse pupillary block. Pavlin and coworkers44 have shown that accommodation can increase iris concavity in some patients with pigment dispersion syndrome, most likely through an accommodation-induced relative increase in anterior chamber pressure secondary to forward movement of the lens surface. They found that laser iridotomy could be used to prevent these changes in iris configuration seen with accommodation. Finally, exercise may augment posterior iris bowing, possibly through a heightened ocular pulse, which may increase cyclic aqueous movements through the pupil.45 As with accommodation, the posterior iris bowing that occurs with exercise in these patients can be eliminated by laser iridotomy
What Is the Clinical Course of Pigmentary Glaucoma?
The onset of pigment dispersion syndrome is thought to occur in most patients during the third decade of life. An active phase of pigment dispersion, which may last for many years, is typically followed by a regression phase, during which pigment liberation is markedly reduced or ceases completely. During this later phase, decreasing corneal endothelial pigment, a reduction in the number of iris transillumination defects,38 and normalization of IOP46 are often seen clinically. Older patients may be left with only a pigment reversal sign, in which the normal pattern of pigmentation reverses, becoming darker superiorly than inferiorly, suggesting a previous history of pigment dispersion.47
Studies have shown that most patients with pigment dispersion syndrome do not develop increased IOP and optic nerve damage.11,19,20 Richter and associates20 reported that in most patients with pigmentary glaucoma, active pigment dispersion was associated with elevated IOP and progression of glaucomatous nerve damage. Küchle and co-workers48 have recently developed a technique using the laser flare-cell meter to quantify aqueous melanin granules in pigmentary glaucoma patients, which may prove useful in evaluating eyes with pigment dispersion syndrome and assessing treatment efficacy.
What Causes Elevated IOP in Pigmentary Glaucoma Patients?
Richardson et al49 have shown that in eyes with pigmentary glaucoma, trabecular meshwork cells phagocytose dispersed pigment particles and then disintegrate or migrate away from the trabecular beams. This response is typically accompanied by localized collapse of the trabecular meshwork, with resultant decreased aqueous outflow facility. In patients with pigment dispersion syndrome, however, the intertrabecular spaces remain open and, except for the presence of pigment-laden epithelial cells, meshwork architecture remains intact. Campbell and co-workers50 performed perfusion studies on rhesus monkey eyes and found that fewer particles could produce an acute rise in IOP than would be expected with intertrabecular obstruction alone. Histologic studies on those monkeys with acute IOP elevation from pigment perfusion showed features similar to those seen in POAG, such as collapse of intertrabecular spaces associated with obstruction by debris.50 From these results the authors concluded that trabecular meshwork damage in pigmentary glaucoma arises from some other mechanism in addition to intertrabecular obstruction.
What Is the Differential Diagnosis of Pigmentary Glaucoma?
The differential diagnosis of pigmentary glaucoma is listed in Table 9–3. Patients with pseudoexfoliation syndrome typically develop peripupillary iris atrophy in contrast to the midperipheral iris transillumination defects seen exclusively in pigment dispersion syndrome and pigmentary glaucoma. Although small amounts of pigment may collect on the corneal endothelium in pseudoexfoliation syndrome, a typical Krukenberg’s spindle does not develop. The trabecular pigmentation in pseudoexfoliation syndrome is patchy and irregular as opposed to the dense homogeneous band found in pigmentary glaucoma. Finally, pigmentary glaucoma patients lack the typical collection of pseudoexfoliative material on the anterior lens capsule seen in pseudoexfoliation syndrome.
Pigmentary glaucoma can be readily distinguished from uveitis by its lack of other signs of inflammation such as the formation of posterior or peripheral anterior synechiae and keratic precipitates. Ocular melanosis (congenital melanosis oculi) is typically unilateral and consists of abnormal pigmentation in the sclera, episclera, and uveal tissue. Signs of pigmentary glaucoma are absent. Placement of a posterior chamber intraocular lens in the ciliary sulcus rather than the capsular bag following cataract extraction may result in chronic iris chafing by the lens haptics with pigment liberation and secondary glaucoma.51
Unilateral pigmentary dispersion syndrome with secondary glaucoma is typically seen in association with an iris or ciliary body cyst or melanoma. Rarely, iris ring melanoma may masquerade as unilateral pigmentary glaucoma.52 Other signs of pigmentary glaucoma, such as transillumination defects or Krukenberg’s spindles, are typically absent in these patients, whereas the cyst or tumor can often be found easily during gonioscopy or on dilated examination. Unilateral pigment dispersion with secondary glaucoma may also be seen with angle recession.53 Evidence on gonioscopy or history of ocular trauma is typically present. The decision tree for the differential diagnosis of patients with pigmentary glaucoma is shown in Figure 9–1.
What Retinal Disorders May Be Associated with Pigmentary Glaucoma?
Retinal abnormalities have been reported in patients with pigment dispersion syndrome and pigmentary glaucoma (Table 9–4). Chew and Deutman36 initially described retinal pigment epithelial (RPE) changes in the posterior pole of a patient with pigment dispersion syndrome. Fluorescein angiography showed a “fishnet” pattern typical of pigmented pattern dystrophy of the RPE. Cardillo Piccolino and associates37 later described two brothers with pigmentary glaucoma who presented with bilateral degeneration of the peripapillary RPE, one of whom developed recurrent serous RPE detachments and neovascular choroidal membranes. Weseley and co-workers54 reported an incidence of lattice degeneration of the peripheral retina of 16.0% in 119 pigment dispersion patients. Scuderi and co-workers55 found lattice degeneration to be present in 33.3% of 24 patients with pigment dispersion. The authors accounted for the higher incidence of lattice degeneration in their series compared to that of Weseley et al14 by noting that the mean age of their study group was approximately 10 years higher than Weseley’s and that their patients were less myopic and thus less likely to exhibit lattice degeneration, which is more common in patients with myopia of 3.00 diopter (D) or less.56 Weakness of the peripheral retina was also noted by Brachet and Chermet,57 who reported 19 cases of rhegmatogenous retinal detachment in pigmentary dispersion patients. Finally, Scheie and Cameron8 found an incidence of retinal detachment of 6.6% in pigment dispersion patients and 7.6% in pigmentary glaucoma patients. This is much higher than the expected annual incidence of phakic, nontraumatic retinal detachment in the general population, which has been reported to be 0.005% to 0.01%. Furthermore, the majority of patients with retinal detachments in Scheie and Cameron’s series were only moderately myopic, suggesting that the increased risk for retinal detachment in pigment dispersion syndrome is substantial.
Pseudoexfoliation syndrome |
Uveitis |
Ocular melanosis |
Pseudophakic pigment dispersion |
Intraocular cyst or tumor |
Angle recession with pigment dispersion |
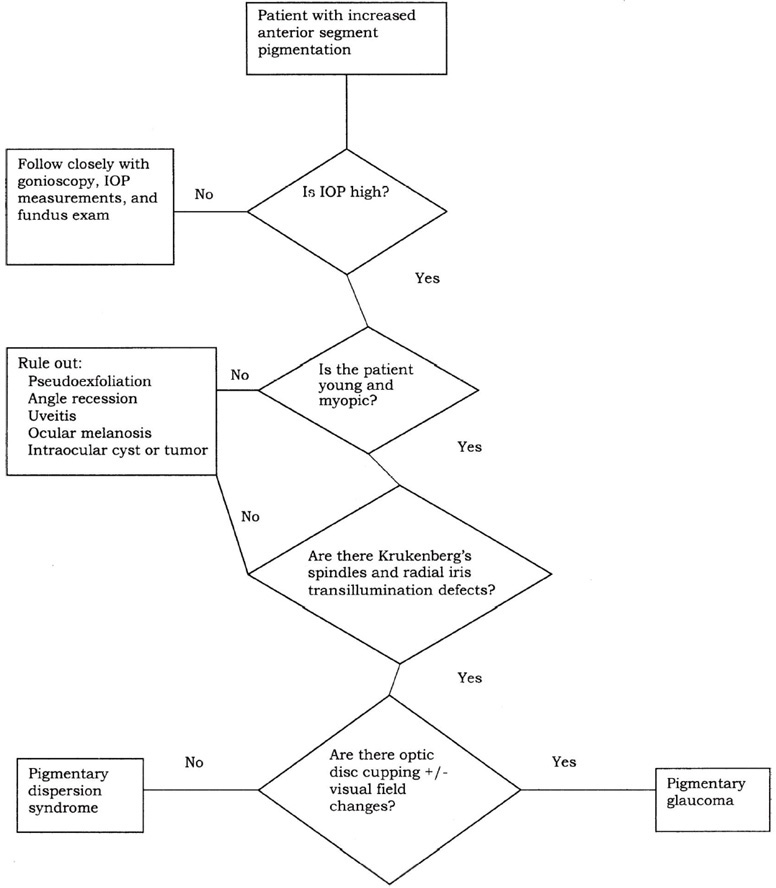
Figure 9-1. Differential diagnosis of patient with pigmentary glaucoma.
Degeneration of the retinal pigment epithelium |
Lattice degeneration |
Increased risk of rhegmatogenous retinal detachment |
Treatment and Management
What Is the Treatment for Pigmentary Glaucoma?
The treatment for pigmentary glaucoma is similar to that for POAG (Table 9–5). Often with mild IOP elevation and optic disc damage, a topical beta-blocker is sufficient to lower IOP and prevent further visual field loss. When IOP remains elevated or evidence of visual field progression develops, other aqueous suppressants, such as α2-agonists or carbonic anhydrase inhibitors, may be added. Because the conventional aqueous outflow pathway is compromised in pigmentary glaucoma, drugs that increase outflow through the uveoscleral pathway, such as prostaglandin F2α, may have added beneficial effect.
Aqueous suppressants |
Beta-blockers |
α2-agonists |
Carbonic anhydrase inhibitors |
Increased aqueous outflow |
Prostaglandin F2α |
Miotics |
Pilocarpine |
Carbachol |
Dapiprazole |
Argon laser trabeculoplasty |
Laser iridotomy |
Filtration surgery |
In some patients, the addition of a miotic, such as pilocarpine or carbachol, may be necessary. Along with increasing aqueous outflow, miotics may help prevent progression of the disease by flattening the iris and preventing further iridozonular contact and pigment dispersion.28 Unfortunately, miotic therapy is often poorly tolerated in young myopic patients because of accommodative spasm and increased myopia. In these patients, sustained-release forms of the drug, such as pilocarpine gel or Ocuserts, can sometimes be used more effectively. Miotics should be used cautiously in these patients and only after a dilated retinal exam, because of the association of peripheral retinal pathology and pigmentary glaucoma.57 Because of their ability to produce miosis and iris flattening without ciliary spasm and induced myopia, α-adrenergic antagonists, such as thymoxamine, may someday play an important role in the treatment of pigmentary glaucoma. Mastropasqua and associates58 have shown that long-term therapy with dapiprazole, an α-adrenergic antagonist, results in significantly increased outflow facility in pigmentary glaucoma patients. The disadvantages of dapiprazole include high cost of the medication and short shelf-life after reconstituting.
For those patients who are not adequately controlled with medical therapy, argon laser trabeculoplasty (ALT) can sometimes be used successfully, especially in younger patients, although its effect is usually short-lived. Lunde59 reported that an initial drop of 10.6 mm Hg following ALT in 13 eyes of 10 patients with pigmentary glaucoma was followed by an increase in IOP to higher than baseline levels after 9 months. Lehto60 reported an initial IOP-lowering effect of 53% in pigmentary glaucoma patients, which decreased to only 14% after 3 months. When performing ALT in pigmentary glaucoma patients, lower energy levels should be used because of the increased pigmentation and energy absorption of the trabecular meshwork.
Peripheral iridotomy has been shown to alleviate the reverse papillary block and iris concavity seen in pigment dispersion patients, thus preventing iridozonular contact and further pigment dispersion. Gandolfi and Vecchi61 found a markedly reduced incidence of ocular hypertension in eyes with pigment dispersion syndrome following laser iridotomy. At this point, the role that laser iridotomy may play in the management of pigment dispersion syndrome and pigmentary glaucoma requires further study.
Filtration surgery is necessary in those patients who fail medical and laser therapy. Scheie and Cameron8 found that pigmentary glaucoma patients more frequently required surgical intervention than patients with POAG. Success rates of filtration surgery for these two groups of patients are similar.
Future Considerations
Where Is the Research on Pigmentary Glaucoma Heading?
Much has been learned about pigmentary glaucoma since its original description by Sugar and Barbour4 in the 1940s. Once thought to be a rare disorder, it is now known to be fairly common, with a prevalence of 2.45% in a large screening population.16 Future considerations for the diagnosis and treatment of pigment dispersion syndrome and pigmentary glaucoma appear quite promising. The α-adrenergic antagonists such as dapiprazole and thymoxamine, which produce miosis and iris flattening with increased aqueous outflow facility and without the induced myopia and ciliary spasm of pilocarpine, are promising, α-adrenergic antagonists, however, are not yet available for clinical use in the United States. Peripheral iridotomy, with its ability to alleviate the reverse pupillary block mechanism described by Karickhoff40 may also become more important in the treatment of the disorder, especially early in its course. New methods of patient evaluation, such as the technique for quantification of aqueous melanin granules as described by Küchle and associates48 may help us better assess treatment efficacy. Finally, gene therapy may someday play a role in the disorder as we build on the work of Andersen et al15 and better characterize the genetic basis of pigment dispersion syndrome.
References
2. Von Hippel E: Zur pathologischen Anatomie des Glaukom. Arch Ophthalmol 1901;52:498.
3. Jess A: Zur Frage des Pigmentglaukoms. Klin Monatsbl 1923;71:175.
4. Sugar HS, Barbour FA: Pigmentary glaucoma: a rare clinical entity. Am J Ophthalmol 1949;32:90–92.
5. Bick MW: Pigmentary glaucoma in females. Arch Ophthalmol 1957;58:483–494.
7. Sugar HS: Pigmentary glaucoma: a 25-year review. Am J Ophthalmol 1966;62:499–507.
10. Weber PA, Dingle JB: Pigmentary glaucoma in a black albino. Ann Ophthalmol 1983;15:454–455.
17. Mapstone R: Pigment release. Br J Ophthalmol 1981;65:258–263.
18. Wilensky JT, Buerk KM, Podos SM: Krukenberg’s spindles. Am J Ophthalmol 1975;79:220–225.
24. Calhoun FP Jr: Pigmentary glaucoma and its relation to Krukenberg’s spindles. Am J Ophthalmol 1953;36:1398–1415.
25. Lichter PR, Shaffer RN: Iris processes and glaucoma. Am J Ophthalmol 1970;70:905–911.
46. Speakman JS: Pigmentary dispersion. Br J Ophthalmol 1981;65:249–251.
56. Byer NE: Lattice degeneration of the retina. Surv Ophthalmol 1979;23:213–247.
57. Brachet A, Chermet TM: Association glaucome pigmentaire et de-collement de retine. Ann Ocul (Paris) 1974;207:451–57.
61. Gandolfi SA, Vecchi M: Effect of a YAG laser iridotomy on intraocular pressure in pigment dispersion syndrome. Ophthalmology 1996;103:1693–1695.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


