Chapter 132 Phakomatoses
Introduction
The phakomatoses are a group of syndromes characterized by systemic hamartomas of the eye, brain, skin, and sometimes the viscera and bones.1–4 As a result of the multisystem involvement, the phakomatoses are also known as oculoneurocutaneous syndromes. Variable clinical manifestations of these syndromes are recognized and comprehensive care of the patient often involves coordination of many specialists.
The term “phakoma” was first used by Van der Hoeve in 1932 to indicate a mother spot, or birthmark, a characteristic finding in many of these entities.4 At that time, retinal and cerebellar hemangiomatosis (von Hippel–Lindau syndrome), neurofibromatosis (von Recklinghausen syndrome), and tuberous sclerosis complex (Bourneville syndrome) were all classified as phakomatoses. Later, encephalofacial hemangiomatosis (Sturge–Weber syndrome), racemose angiomatosis (Wyburn-Mason syndrome), and cavernous hemangioma of the retina with cutaneous and central nervous system involvement were included with these conditions. More recently, other entities such as organoid nevus syndrome and oculocutaneous melanocytosis have been categorized with these classic oculoneurocutaneous syndromes.
Definition of hamartia, hamartoma, chorista, choristoma
To better understand the phakomatoses, certain terms such as hamartia, hamartoma, chorista, and choristoma should be defined.1 Hamartia and hamartoma are terms that refer to a malformation that is composed of tissues normally present at the location where it develops. A hamartia is a nontumorous anomaly and a hamartoma is a tumorous malformation. The term systemic hamartomatosis is used to designate multiple organ involvement. Examples of hamartomas include the retinal capillary hemangioma (hemangioblastoma) that occurs from already present vascular tissue in the retina in von Hippel Lindau syndrome, or the cutaneous peripheral nerve tumors that occur from the already present neural tissue within the skin in patients with neurofibromatosis.
Most of the tumors that develop with the phakomatoses are benign. They are usually stationary or slowly progressive lesions that generally lack the capacity for limitless proliferation found with cancers.1 Some phakomatoses can be associated with malignant neoplasms. For example, there is an increased incidence of malignant schwannomas of the peripheral nerves in patients with neurofibromatosis. Renal cell carcinoma occurs with greater frequency in patients with von Hippel Lindau disease.
Neurofibromatosis (von recklinghausen syndrome)
Neurofibromatosis is an oculoneurocutaneous syndrome characterized by multisystem involvement.5,6 It is transmitted by an autosomal dominant mode of inheritance with about 80% penetrance. About one-half of the cases occur initially as spontaneous mutations with a negative family history.
Neurofibromatosis type 1
General considerations
Neurofibromatosis type 1 occurs at a rate of 1 in 3000 persons, but it is estimated that the frequency could be higher as some individuals manifest only mild features. Approximately one-half of affected patients represent a new mutation. This condition is caused by an autosomal dominant mutation in the NF1 gene that leads to decreased production of the protein neurofibromin, which has a tumor suppressor function. Only one deleted is necessary to manifest this condition. The NF1 gene is on long arm of chromosome 17. There have been more that 250 mutations identified. Complete gene deletion leads to severe phenotype. This highly penetrant phenotype has a wide variety of manifestations and can vary within families. Another locus, the SPRED1 gene, has been found in patients with “mild neurofibromatosis” and this represents Legius syndrome.7
The criteria for diagnosis of neurofibromatosis type 1 are listed in Table 132.1.8,9 This condition more often affects patients of the Caucasian race and equally in boys and girls. Scoliosis can be a prominent feature.
Table 132.1 Diagnostic criteria for neurofibromatosis type 1 (NF1) – at least two of the following seven criteria should be present for diagnosis*
| Feature | |
|---|---|
| 1.Café au lait | ≥6 café au lait spots larger than 5 mm diameter in prepubertal children (<10 years) |
| or | |
| ≥6 café au lait spots larger than 15 mm in diameter in postpubertal individuals (adults) | |
| 2.Freckles in axilla or inguinal region | Crowe sign |
| 3.Skin neurofibroma | ≥2 typical neurofibroma |
| or | |
| ≥1 plexiform neurofibroma | |
| 4.Optic nerve glioma | |
| 5.Iris Lisch nodules | ≥2 lesions |
| 6.Osseous lesion | Sphenoid dysplasia |
| or | |
| Long bone abnormalities (cortex thinning or pseudoarthrosis) | |
| 7.Relative (1st degree) with NF1 by above criteria | Parent, sibling or offspring |
* Some manifestations do not appear until later life, delaying diagnosis. (Adapted from Stumpf DA, Alksne JF, Annegers JF. Neurofibromatosis. Conference statement. National Institute of Health Consensus Development Conference. Arch Neurol 1988; 45:575–88 and Gutmann DH, Aylsworth A, Carey JC, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997;278:51–7.9)
Ophthalmologic features
Neurofibromatosis has the most diversified ocular findings among the phakomatoses.10–17 This condition can involve the eyelid, conjunctiva, aqueous outflow channels, uveal tract, retina, orbit, and optic nerve (Fig. 132.1).
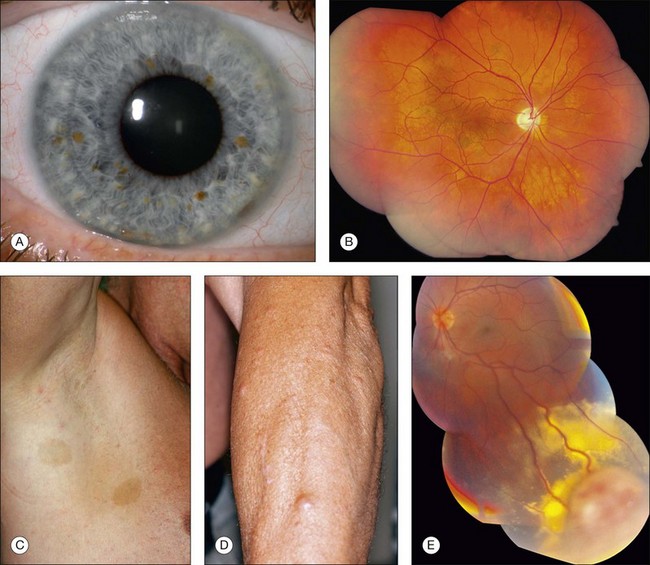
Eyelid involvement is characterized by nodular or plexiform neurofibroma. Nodular neurofibroma appears as a solitary or multifocal painless, smooth-surfaced and well-defined mass, often the size of a pea, and without color change. Plexiform neurofibroma presents as a diffuse thickening of the eyelid that can produce the typical S-shaped curvature to the eyelid, a finding highly characteristic of neurofibromatosis. The conjunctiva can be involved by diffuse or localized neurofibromas. Patients with neurofibromatosis have an increased incidence of congenital glaucoma, which can be secondary to several mechanisms.10 There is a high association of neurofibromatosis type 1 with optic nerve glioma.12
A variety of ocular fundus lesions can occur in patients with neurofibromatosis. Multiple iris hamartomas, known as Lisch nodules, are the most common uveal abnormality.11 Iris Lisch nodules are the most common ophthalmic abnormality of neurofibromatosis type 1. These characteristically orange-tan nodules appear in early childhood (usually by age 6 years) as discrete, multiple, bilateral tumors of the anterior border layer of the iris, classically measuring less than 1 mm diameter and best detected by slit lamp biomicroscopy. Histopathologically, iris Lisch nodules are hamartomas composed of aggregates of melanocytes on the anterior border layer of the iris.
The choroidal findings in patients with neurofibromatosis type 1 include unifocal or multifocal choroidal nevus, diffuse plexiform neurofibroma, neurilemoma, and melanoma. Multiple bilateral, choroidal nevi are highly suggestive of neurofibromatosis type 1. They are usually small, ill-defined, and randomly distributed. Choroidal neurofibroma usually appears as a diffuse thickening of the uveal tract from an increased number of neurofibromatous and melanocytic elements. Choroidal neurilemoma (Schwannoma) is a rare finding and manifests as a circumscribed, amelanotic elevated tumor.14 Most solitary choroidal neurilemomas, however, are not associated with neurofibromatosis. There appears to be a higher incidence of uveal melanoma in patients with neurofibromatosis.15,16
Several retinal and optic disc lesions can occur with neurofibromatosis. Retinal astrocytic hamartoma is a manifestation of neurofibromatosis, but is more common with tuberous sclerosis complex. Retinal vasoproliferative tumor can occur with neurofibromatosis, leading to exudative retinopathy and risk for blindness.17 Fundus changes can occur secondary to optic nerve glioma including optic disc edema, optic atrophy, opticociliary shunt vessels and, rarely, central retinal vein obstruction.
Neurofibromatosis type 2
General considerations
Neurofibromatosis type 2 is a multisystem disorder with prominent features of central nervous system tumors including bilateral vestibular schwannomas (acoustic neuromas), spinal cord schwannomas, meningiomas, gliomas, and juvenile posterior subcapsular cataract. This condition is also referred to as MISME syndrome, a mnemonic referring to related tumors of MIS (multiple inherited schwannomas), M-meningiomas, and E-ependymomas.18 Cutaneous features are less often seen with this form of neurofibromatosis. The criteria for diagnosis of neurofibromatosis type 2 are listed in Table 132.2.
Table 132.2 Diagnostic criteria for neurofibromatosis type 2 (NF2). Diagnosis is established with at least one of the three items listed below
Feature 1 Bilateral 8th cranial nerve tumors confirmed on magnetic resonance imaging or computed tomography |
(Adapted from Stumpf DA, Alksne JF, Annegers JF. Neurofibromatosis. Conference statement. National Institute of Health Consensus Development Conference. Arch Neurol 1988;45:575–88 and Gutmann DH, Aylsworth A, Carey JC, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997;278:51–7.9)
Neurofibromatosis type 2 can be associated with reduced life span secondary to central nervous system tumors, particularly if they are present at a young age and are multiple. The average age at symptom onset is approximately 20 years but can be delayed. Early age at symptoms and the presence of intracranial meningioma at diagnosis are two signs of higher risk for disease severity and mortality. In an analysis of 150 affected patients in 1992, more than 40% were expected to die by 50 years.19 Recent advances in treatment have extended life prognosis.
The incidence of neurofibromatosis type 2 is 1 in 25 000 live births and has nearly 100% penetrance by 60 years of age.20 It is estimated that this condition has a diagnostic prevalence of 1 in 100 000 people in 2005. Neurofibromatosis type 2 is related to a mutation in the NF2 gene at chromosome 22q12.2. This gene produces merlin (also called neurofibromin-2), a tumor suppressor. When mutated, decreased function of merlin leads to the uncontrolled development of tumors, particularly in the central nervous system. One-half of affected patients have a de novo mutation.
Ophthalmologic features
Neurofibromatosis type 2 displays three important ophthalmologic findings, notably posterior subcapsular cataract in childhood, combined hamartoma of the retina and retinal pigment epithelium, and epiretinal membranes (Table 132.3). The juvenile posterior subcapsular cataract (<50 years) is a criterion for diagnosis of this condition. Other lens opacity in the capsular or cortical region of young patients are believed related to neurofibromatosis type 2. Lisch nodules are not a feature of neurofibromatosis type 2.
Table 132.3 Frequency of clinical features in patients with neurofibromatosis type 2
| Feature | Frequency associated with neurofibromatosis type 2 (%) |
|---|---|
| Ophthalmologic features | |
| Juvenile posterior subcapsular cataracts | 60–81 |
| Epiretinal membranes | 12–40 |
| Retinal hamartomas | 6–22 |
| Cutaneous features | |
| Cutaneous tumors | 59–68 |
| Cutaneous plaques | 41–48 |
| Subcutaneous tumors | 43–48 |
| Intradermal tumors | Rare |
| Central nervous system features | |
| Bilateral vestibular (cranial nerve 8) schwannomas | 90–95 |
| Other cranial nerve schwannomas | 24–51 |
| Intracranial meningiomas | 45–58 |
| Spinal tumors | 63–90 |
| Extramedullary | 55–90 |
| Intramedullary | 18–53 |
| Peripheral neuropathy | 66 |
(Adapted from Asthagiri AR, Butman JA, Kim HJ, et al. Neurofibromatosis type 2. Lancet 2009;373:1974–86.20)
Epiretinal membranes and combined hamartoma of the retina and retinal pigment epithelium can have overlapping clinical phenotype and can be multifocal. Of those with severe clinical features of neurofibromatosis type 2, 80% display epiretinal membranes.21 The hamartomas are along the inner retina but lead to prominent retinal dragging, corkscrew retinal vessels, gray-green appearance and tumor formation.22,23
Management
Patients with neurofibromatosis type 2 should have annual ophthalmic, neurologic, dermatologic, and auditory examinations. This requires a multidisciplinary team. Surgical resection of symptomatic neurologic tumors is performed, but radiotherapy or chemotherapy can be used, particularly for ependymomas. Erlotinib has been used for unresectable progressive vestibular schwannomas and this medication is under trial. Additionally, bevacizumab and Gleevec have been investigated for treatment of schwannomas. Regarding ophthalmic care, cataract surgery can be beneficial. Additionally, monitoring of epiretinal membrane with clinical examination and optical coherence tomography, with surgical removal if progressive, can be considered.22
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


