Chapter 65 Phakomatoses
Neurofibromatoses
Neurofibromatosis type 1
NF1 is a progressive disease and the final manifestations are extremely variable. Existing lesions tend to enlarge gradually and new lesions develop. The National Institutes of Health (NIH) diagnostic criteria are listed in Box 65.1.
Box 65.1
NIH diagnostic criteria for NF1
The diagnostic criteria are met if two or more of the following are found:
1. Six or more café-au-lait macules over 5 mm in greatest diameter in prepubertal individuals and over 15 mm in greatest diameter in postpubertal individuals
2. Two or more neurofibromas of any type or one plexiform neurofibroma
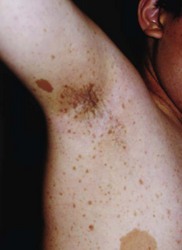
3. Freckling in the axillary or inguinal regions
5. Two or more Lisch nodules (iris hamartomas)
6. A distinctive osseous lesion such as sphenoid dysplasia or thinning of long bone cortex
Genetics
NF1 is the most common single gene disorder affecting the nervous system, occurring in approximately 1 in 3000 people. Inheritance is autosomal dominant with 100% penetrance, but highly variable expressivity. The spontaneous mutation rate is up to 42%.1
The protein associated with NF1, neurofibromin, down regulates the rat sarcoma viral oncogene homolog (RAS)−mitogen activated protein kinase (MAPK) pathway. Unopposed Ras activity leads to cell growth and activation of downstream signaling intermediates, including the mammalian target of rapamycin (mTOR) protein.2 NF1 mutations give rise to malignant tumors consistent with the “two hit” hypothesis: a germ-line mutation is the initial “hit”; neurofibromas arise from Schwann cells that undergo loss of heterozygosity at the NF1 gene through somatic mutations (the second “hit”).
Clinical presentation
Ocular findings are important in the NF1 diagnostic criteria (see Box 65.1), including Lisch nodules, orbital plexiform neurofibroma, sphenoid wing dysplasia, and optic nerve glioma.
Lisch nodules
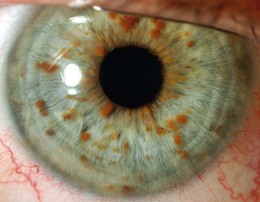
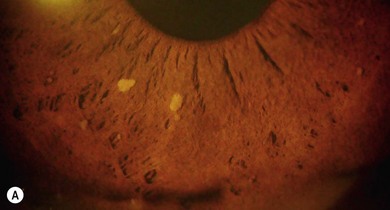
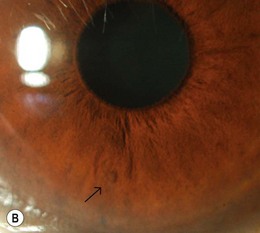
Lisch nodules (melanocytic hamartomas) are dome-shaped, discrete lesions on the anterior surface of the iris or in the angle, usually bilateral. They are usually orange-brown (Fig. 65.1), appearing darker than blue irides but paler than brown irides (Fig. 65.2). Most are round and evenly distributed on the iris. Their size varies from a pinpoint to involvement of a segment of iris, sometimes confluent. In NF1, they are present in one-third of 2.5-year-olds, half of 5-year-olds, three-quarters of 15-year-olds, and almost all adults over 30.3 They occur earlier than neurofibromas and are, therefore, a useful marker for NF1. They need to be distinguished from iris nevi.
Anterior segment and uvea
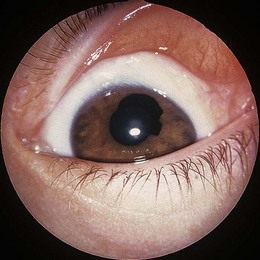
Rarely, congenital glaucoma with buphthalmos occurs in NF1, often associated with an ipsilateral upper lid plexiform neurofibroma. Congenital ectropion uveae (Fig. 65.3), iris heterochromia, angle abnormalities, and posterior embryotoxon may predispose to later onset glaucoma. Cataract is not a feature of NF1. Pigmentary hamartomas (choroidal nevi) may involve the posterior uveal tract in up to 35%.
Skin, lids and orbits
Café-au-lait spots are hyperpigmented macular skin lesions, mostly present at birth and all appear by 1 year, enlarging at puberty. Common on the trunk, they are absent from the scalp, eyebrows, palms, and soles (Fig. 65.4). Histologically, there is melanocytic hyperplasia with increased pigmentation in the basal layer of the epidermis.
Neurofibromas are present in 30–50% of NF1 patients.4 They are network-like tumor growths involving multiple fascicles of nerve(s); four types are recognized (Table 65.1). The presence and growth pattern of neurofibromas is age related; in infancy and early childhood, diffuse plexiform neurofibromas are most active giving rise to cosmetic and visual problems within the orbit. Cutaneous or subcutaneous neurofibromas develop and grow fastest at puberty, the late teens, and during pregnancy. There is a lifelong risk of malignant transformation with poor prognosis for 5-year survival.5
Table 65.1 – Neurofibroma clinical features4
| Neurofibroma types | Location | Clinical signs |
|---|---|---|
| Discrete cutaneous | Epidermis and dermis | Moves with skin, blueish tinge |
| Subcutaneous | Deep to dermis | Skin moves over, firm and rounded feel, located along peripheral nerves |
| Nodular plexiform | Localized interdigitation with normal tissues | “Bag of worms” feel (Figs 65.5, 65.6) |
| Diffuse plexiform | Infiltrate widely and deeply | Smooth, slightly irregular skin thickening |
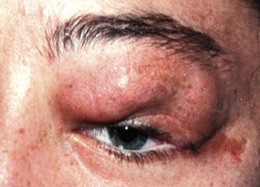
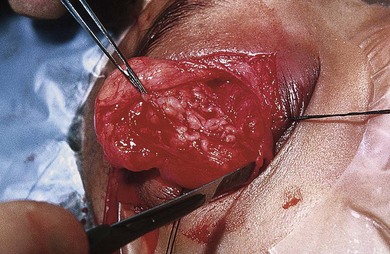
Fig. 65.6 The worm-like consistency of an orbital plexiform neurofibroma can be seen.
(Patient of the University of British Columbia.)
Complete surgical resection of plexiform neurofibromas is difficult due to their large size, location, local invasiveness, or involvement of critical peripheral nerves. Regrowth following subtotal resection is common. Chemotherapy is not effective. Promising clinical trials with biologic agents are underway.6
Skeletal anomalies
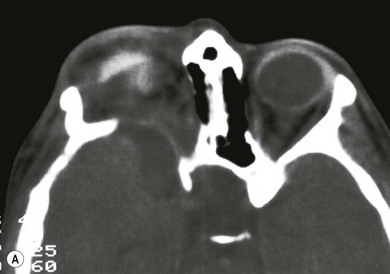
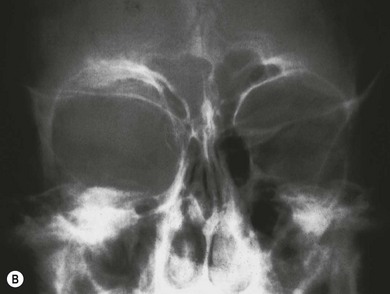
Hypoplasia of the greater and lesser spenoid wings and widening of the superior orbital fissure may occur. Other osseous abnormalities include abnormal vertebrae, scoliosis, pseudoarthrosis of long bones, and subperiosteal changes (Fig. 65.7). Neurofibromas may interfere with cranial nerve function.
Optic disk and central nervous system
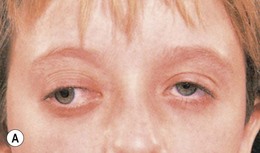
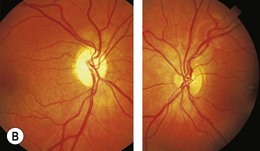
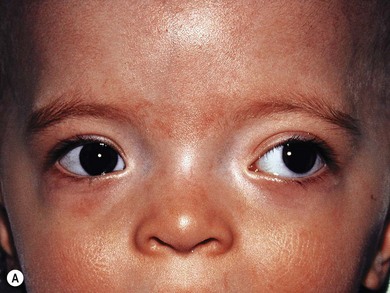
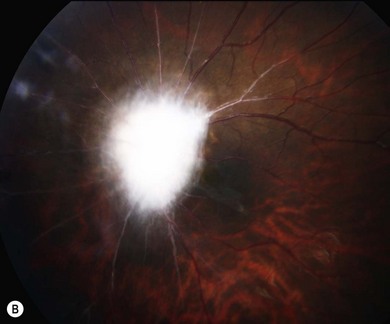
Optic pathway gliomas are a significant cause of visual morbidity (see Chapter 23). Optic disk swelling (Fig. 65.8), strabismus (Fig. 65.9), or pallor suggests optic nerve or chiasmal glioma. Retinal manifestations are rare in NF1. The commonest consequence of NF1 in childhood and a major concern of parents is cognitive impairment. Overt mental retardation is rare, but a wide range of learning disabilities, academic underachievement, and behavioral problems may occur.
Neurofibromatosis type 2
Neurofibromatosis type 2 (NF2) affects 1 in 35 000 live births and is characterized by bilateral vestibular schwannomas, multiple central nervous system (CNS) tumors, cataracts, and retinal abnormalities. The diagnostic criteria for NF2 are shown in Box 65.2. Two patterns of presentation exist. Mild disease presents late (> 25 years), with few tumors other than bilateral vestibular schwannoma, and slow progression. Those with severe disease have onset before age 25, multiple CNS tumors (at least two of one type), and rapid progression, severe handicap or death before reproductive age. Treatment is multidisciplinary because of the complexities of the multiple and progressive lesions.7
Box 65.2
Revised diagnostic criteria (Manchester) for NF2
NF2 may be diagnosed in any one of the following situations:
• A. Bilateral vestibular schwannomas
• B. First-degree family relative with NF2 and either unilateral vestibular schwannoma or any two of: meningioma, schwannoma, glioma, neurofibroma, posterior subcapsular lenticular opacities
• C. Unilateral vestibular schwannoma and any two of: meningioma, schwannoma, glioma, neurofibroma, posterior subcapsular lenticular opacities
• D. Multiple meningiomas (two or more) and unilateral vestibular schwannoma or any two of: schwannoma, glioma, neurofibroma, cataract
Genetics
The NF2 tumor suppressor gene Merlin is localized to the cell membrane–cytoskeletal interface where it indirectly acts through its membrane organization of proteins. Merlin regulates downstream mitogenic signaling pathways. Drugs targeting these pathways (i.e. sorafenib, trastuzumab, lapatinib, LY294002, protein kinase inhibitors, p21-activated kinase inhibitors) and tumor angiogenesis (bevacizumab) are under investigation as potential treatments for NF2.7
Systemic findings
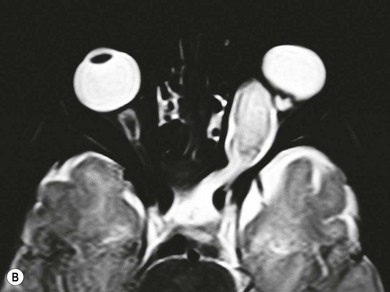
The hallmark of NF2 is bilateral vestibular schwannomas (“acoustic neuromas”) which may only be evident on gadolinium-enhanced MRI (see Fig. 65.11C ). The origin of the trigeminal nerve is another frequent intracranial site for schwannomas; they may occur on multiple cranial nerves. Anti-VEGF (vascular endothelial growth factor) therapy improves hearing and stabilizes tumor growth.8
Ocular findings
Most of the ophthalmic findings in NF2 (see Box 65.2) are congenital but may become more symptomatic over time.9 Visual loss increases morbidity since progressive bilateral hearing loss is so common in this condition.
Anterior segment
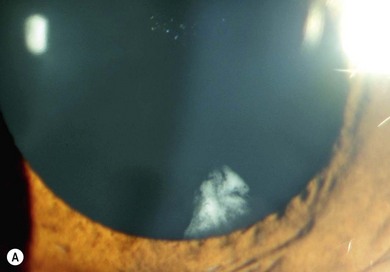
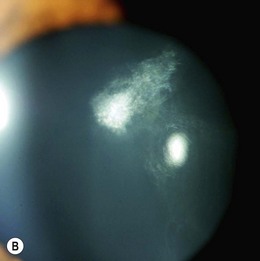
The ocular changes of NF2 are early diagnostic markers. 55–87% of affected patients have presenile central posterior subcapsular lens opacities, usually with little effect on vision (Fig. 65.10A,B). Corneal hypoesthesia from trigeminal nerve schwannoma, and decreased tear production, reduced blinking and lagophthalmos from facial nerve palsy may adversely affect the outcome of cataract surgery.
Posterior segment
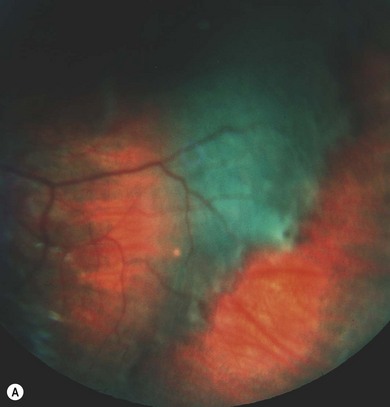
Epiretinal membranes occur in up to 80% of patients with severe NF2. They are translucent, semitranslucent, or whitish gray membranes with prominent whitish edges demarcating their borders. They do not usually cause substantial loss of visual acuity (Fig. 65.11).
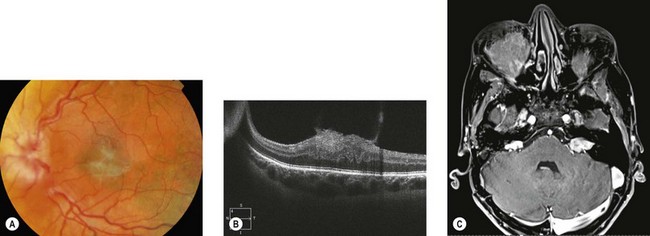
Combined retinal and pigment epithelial hamartomas occur in 6−22% of NF2 patients, especially with nonsense mutations.9 They are slightly raised masses most frequently in the posterior pole (Fig. 65.12), commonly causing reduced vision. Optic nerve sheath meningiomas, sometimes bilateral, can cause visual loss (see Chapter 23). They may be missed on MRI unless fat suppression techniques are used.
Prognosis
Increased understanding of the clinical manifestations and improved precision of genetic tests and imaging studies have improved early diagnosis of patients. Survival is extended because of advances in treatment available at specialized centers and the recognition of mildly affected people with mosaic disease (Box 65.3).



