CHAPTER 165 Peripheral Vestibular Disorders
Clinical Relevance
The lifetime prevalence of vestibular vertigo is 7.4%.1 In 80% of affected persons, vertigo resulted in a medical consultation, interruption of daily activities, or sick leave. Of patients who present to emergency departments with falls of unknown cause, 80% have vestibular impairment, and 40% complain of vertigo.2 Thus, peripheral vestibular disorders constitute a significant medical problem. The most important clinical feature in the diagnosis of peripheral vestibular dysfunction is its pattern of presentation or history.
Historical Background
Before the 1860s, dizziness and balance problems were thought to be exclusively central disorders, often referred to collectively as “cerebral congestion” or lumped in with epilepsy. As early as the 1820s, post-rotation nystagmus was observed in mental patients after rotation in cages as a means to subdue them. Jan E. Purkinje hypothesized that this effect was central in origin. Vertigo symptoms during this period often were treated with leaching, purging, and cupping. Around this time emerged the first hints that equilibrium may have a peripheral component. Pierre Flourens noticed that pigeons would fly in circles in the same orientation as an ablated semicircular canal.3
The existence of peripheral vestibular disorders was proposed by Prosper Meniere in 1861.4 Meniere was the director of a large deaf-mute institution in Paris who probably saw patients in whom both vertigo and hearing loss developed immediately after trauma to the ear, allowing him to conclude that both symptoms have a common inner ear origin.5 To support his conclusion he presented the results of an autopsy of a young girl in whom sudden hearing loss and acute vertigo developed after such injury. On autopsy, her brain was found to be normal but the inner ear was filled with blood. It is ironic that this patient probably had leukemia, and not endolymphatic hydrops. Because of this finding, it was commonly believed well into the 20th century that Meniere’s disease was caused by hemorrhage. Before 1940, “Meniere’s disease” was used as a generic term for any peripheral vertigo, especially if it involved hearing loss. The first insight into the true pathophysiology of Meniere’s disease came a decade after his initial report, with Knapp’s hypothesis that inner ear hydrops was similar to ocular glaucoma.6
Early treatments of peripheral vertigo focused on destruction of the end organ. In 1904 the techniques of both eighth cranial nerve section7 and labyrinthectomy8,9 were described. The concept of drainage of the endolymph was first reported by Portmann in 1926.10 Dandy also proposed selective vestibular nerve sectioning using a suboccipital approach during the 1930s, by which he treated more than 600 patients.11 In this early period these procedures carried a high risk of deafness and facial nerve paralysis, as well as significant mortality rates. It was not until 1938, after examination of nerve section specimens taken from two patients who died in the perioperative period, that Hallpike and Cairns were able to report dilatation of the endolymphatic system in patients with Meniere’s disease. This finding was hypothesized to reflect a disruption of the resorptive mechanism.12 This pathologic feature also was independently observed by Yamakawa.13
Treatment of Meniere’s disease that focuses on reversing endolymphatic hydrops has continued to evolve through the present day, with several variations on the Portmann shunt being proposed, such as transmastoid decompression,14 subarachnoid drainage,15 and cochleosacculotomy.16 All of these procedures have had incomplete success in treating vertigo symptoms and carry a risk of hearing loss. A randomized controlled trial has suggested that these procedures have an efficacy similar to that for the placebo effect of surgery,17 although a more recent analysis of these data has suggested that endolymphatic shunts may have a real effect.18 Accordingly, these procedures continue to evolve and be performed.
BPPV is the most common cause of peripheral vertigo. BPPV was first described by Bárány in 1921, and he attributed the disorder to otolith disease.19 After Bárány’s initial description, scattered contradictory and confusing reports of BPPV appeared in the literature.20 The clinical diagnosis of this disorder was not well defined until Dix and Hallpike described the classic positioning which causes a characteristic nystagmus in 1952.21 Like Bárány, however, they believed it was due primarily to otolith disease. It was noted that the disease could be cured by a chemical labyrinthectomy22 and eighth nerve section.23 Schuknecht observed granular deposits on the cupula of the posterior semicircular canal in temporal bone specimens and proposed the “cupulolithiasis” theory to explain the pathophysiology.24 This theory provides a basis for understanding the disorder, although more recent work has shown that the disorder is more commonly due to free-floating particles in the semicircular canal (“canalithiasis”), rather than cupulolithiasis. Gacek proposed transection of only the posterior ampullary nerve for relief of BPPV, confirming the posterior canal origin.25 In most patients, however, the Epley canalith repositioning maneuver is adequate treatment,26 and no surgery is required.
The most recently described etiologic disorder with peripheral vertigo is superior canal dehiscence.27 Affected patients often exhibit a Tullio phenomenon (nystagmus with loud noise) or Hennebert’s sign (eye movements caused by pressure on the external auditory canal). This dehiscence acts as a third window into the middle cranial fossa, which can be plugged using a middle cranial fossa approach28 or a transmastoid approach.29
Benign Paroxysmal Positional Vertigo
The disorder initially was described in 1921 by Bárány.19 He recognized several of the cardinal manifestations of BPPV, including vertical and torsional components of the nystagmus, the brief duration of the nystagmus, and the fatigability of the nystagmus and vertigo. Bárány did not, however, correlate the onset of the nystagmus with the positioning maneuver. He erroneously concluded that an abnormality in the encoding of head position by the otoliths was responsible for the symptoms and signs that he had noted. In 1952, Dix and Hallpike21 reported this entity in a large group of patients. They described their now well-known maneuver for eliciting the classic pattern of nystagmus and its associated symptoms. They recognized the important features of the nystagmus occurring in BPPV, including its latency, directional characteristics, brief duration despite maintaining the offending head position, reversibility upon returning the patient to a seated position, and fatigability from repeated testing. They correctly identified the offending labyrinth but incorrectly concluded that BPPV results from an otolithic disturbance.
Schuknecht30 reviewed the previous studies of BPPV, including the temporal bone histology of several patients who had experienced the disorder. He noted extensive utricular destruction in these patients and damage to other structures supplied by the anterior vestibular artery. This observation and the failure of electrical stimulation of the utriculus or sacculus to produce discrete patterns of nystagmus in laboratory animals led Schuknecht to conclude that abnormal stimulation of the otoliths was not responsible for the disorder. He suggested that the posterior canal crista was the probable source of the dysfunction. According to Schuknecht, BPPV was caused by loose otoconia from the utricle that, in certain positions, displaced the cupula of the posterior canal. Deposition of otoconia on the cupula of the posterior canal, known as “cupulolithiasis,” was later proposed by Schuknecht as the mechanism of BPPV.24 However, this mechanism poorly explained the brief duration of the nystagmus and the reversal of the nystagmus on return to a sitting position.
The suggestion that the mechanism of BPPV could result from deflection of the posterior canal cupula by the movement of debris in the posterior canal was revisited by Hall and colleagues.31 These investigators suggested that the nonfatigable form was due to fixed deposits on the cupula, whereas the fatigable form was attributed to the motion of free-floating material within the lumen of the posterior semicircular canal. In most cases, BPPV-associated nystagmus is most commonly due to motion of material in the lumen of the posterior canal.
Incidence
BPPV is the most common cause of vertigo in patients seen by the otolaryngologist, accounting for 20% to 40% of cases of peripheral vestibular disease32—making it almost twice as frequent as Meniere’s disease. The incidence is difficult to estimate because of the benign, typically self-limited course of the disease. It is thought to range from 10.7 per 100,000 to 17.3 per 100,000 population in Japan33 and has been reported as 64 per 100,000 in a population study from Minnesota.34
The mean age at onset is in the fourth and fifth decades, but BPPV also may occur in childhood.35 In a study of pediatric patients with BPPV, an association with migraine was suggested, leading to the hypothesis that migraine-induced ischemia may be responsible for the release of otoconia in some cases.36 Overall, the incidence increases with age,37 and BPPV is twice as common in women.35
Diagnosis
History
Symptoms occur suddenly and last on the order of seconds but never in excess of a minute.35 The subjective impression of attack reported by the patient frequently is longer.
Episodes of vertigo frequently are clustered in time and separated by remissions lasting months or more. The patient also may report that periods of active disease may be associated with constant feelings of lightheadedness, worsened by head movement.38 These chronic balance problems may be worse on awakening.
In most cases of BPPV, no specific etiologic disorder can be identified. In a large survey by Baloh and colleagues, no cause was identified in 48% of cases.35 The most common known cause was closed head injury, followed by vestibular neuritis. In our experience, BPPV will eventually develop in nearly 15% of patients suffering from vestibular neuritis. Other cited predisposing events include infections and certain surgical procedures, including stapedectomy35 and insertion of a cochlear implant.39 Prolonged bed rest and Meniere’s disease also are predisposing factors.
Findings on Examination
The diagnosis of BPPV is made by observing the classic eye movements in association with the Dix-Hallpike maneuver, combined with a suggestive history. The Dix-Hallpike maneuver is carried out as follows (Fig. 165-1). The patient is positioned supine on an examination table with the head extending over the edge. The patient is lowered with the head supported and turned 45 degrees to one side. The eyes are carefully observed. If no abnormal eye movements are seen, the patient is returned to the upright position. The maneuver is repeated with the head turned in the opposite direction and finally with the head extended supine.40
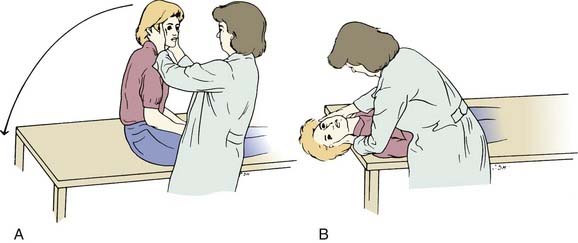
The pattern of response consists of the following: (1) The nystagmus is a combined vertical up-beating and rotary (torsional) component beating toward the downward eye (the superior poles of the eyes beat toward the downward ear). Pure vertical nystagmus is not BPPV.35 (2) A latency of onset of nystagmus (seconds) is common. (3) Duration of nystagmus is short (less than 1 minute). (4) Vertiginous symptoms are invariably associated. (5) The nystagmus disappears with repeated testing (i.e., it is fatigable). (6) Symptoms often recur with the nystagmus in the opposite direction on return of the head to the upright position.38
Canalithiasis of the posterior semicircular canal is the most frequent cause of BPPV. Posterior canal BPPV rarely may be bilateral.41 If the head is not positioned correctly during testing (i.e., not positioned with the head in the plane of the posterior canal during testing of the unaffected side), debris on the affected side may come to rest against the cupula, simulating an excitatory nystagmus from the unaffected ear.42 The lateral semicircular canal has been identified as the affected structure in approximately 12% of cases, with a significant fraction of these being induced by the repositioning maneuver for posterior canal BPPV.43 Lateral canal BPPV can be detected by a variation of the Dix-Hallpike maneuver: The patient is first brought to the supine position with the head resting on (not hyperextended below) the examining table. The head is then turned rapidly to the right so that the patient’s right ear rests on the table. Eye movements are monitored with Frenzel lenses for 30 seconds. The patient is then returned to the supine position (looking upward) and the head is turned rapidly to the left so that the left ear rests on the table. Eye movements are again monitored.
The nystagmus with lateral canal BPPV is horizontal and may beat toward (geotropic) or away from (ageotropic) the downward ear. It often begins with a shorter latency, increases in magnitude while maintaining the test position, and is less susceptible to fatigue with repetitive testing than the vertical torsional nystagmus of posterior canal BPPV. The increased amplitude and duration of the horizontal nystagmus may reflect action of the central velocity storage mechanisms (which perseverate signals from the vestibular periphery, especially those arising from the lateral canal). Cupulolithiasis, arising either independently or in combination with canalithiasis, is more likely to be involved in the etiology of lateral canal BPPV than is the case for posterior canal BPPV. Cupulolithiasis can be differentiated from canalithiasis by minimal or absent latency to the onset of nystagmus. Cupulolithiaisis also can persist for minutes or even as long as the patient remains in the provocative position.44 If the nystagmus is geotropic, the particles are likely to be in the long arm of the lateral canal relatively far from the ampulla. If the nystagmus is ageotropic, the particles may be in the long arm relatively close to the ampulla or on the opposite side of the cupula, either floating within the endolymph or embedded in the cupula.
The superior semicircular canal is affected in only 2% of cases of BPPV.45 The nystagmus expected in cases of superior canal BPPV would be downbeat and torsional. Such a nystagmus can be elicited by standard Dix-Hallpike positioning testing. The right posterior canal lies in roughly the same plane as that of the left superior canal, and vice versa. Placement of the head into the right Dix-Hallpike position will therefore bring the left superior and right posterior canals into the plane of gravity. A nystagmus resulting from the left superior canal BPPV would, in this position, be expected to be downbeat, with a torsional component directed such that the superior part of each eye beats toward the upward ear. Note that this nystagmus is opposite in vertical and torsional direction from that resulting from right posterior canal BPPV.
Test Results
The bedside Dix-Hallpike test21 combined with an appropriate history is key to making the diagnosis. Standard electro-oculography (EOG) and the many videonystagmography devices do not record the torsional eye movements associated with BPPV. The eye movement tracings obtained with these standard devices used for clinical testing reflect solely the associated vertical and horizontal components of the eye movements. The observation of these patterns may be facilitated by the use of Frenzel lenses.
Treatment with Repositioning
First-line therapy for BPPV is organized around repositioning maneuvers that, in cases of canalithiasis, use gravity to move canalith debris out of the affected semicircular canal and into the vestibule. For posterior canal BPPV, the maneuver developed by Epley26 is particularly effective (Fig. 165-2). The maneuver begins with placement of the head into the Dix-Hallpike position, to evoke vertigo. The posterior canal on the affected side is in the earth-vertical plane with the head in this position. After the initial nystagmus subsides, a 180-degree roll of the head (in two 90-degree increments, stopping in each position until any nystagmus resolves) to the position in which the offending ear is up (i.e., the nose is pointed at a 45-degree angle toward the ground in this position) is performed. The patient is then brought to the sitting upright position. The maneuver is likely to be successful when nystagmus of the same direction continues to be elicited in each of the new positions (as the debris continues to move away from the cupula). The maneuver is repeated until no nystagmus is elicited. In our experience, when administered in this way, the Epley maneuver is effective in more than 90% of cases in eliminating BPPV. Medications are not usually given, but a low dose of meclizine or a benzodiazepine 1 hour before may be appropriate if the patient is unusually anxious or susceptible to nausea and vomiting with vestibular stimulation.
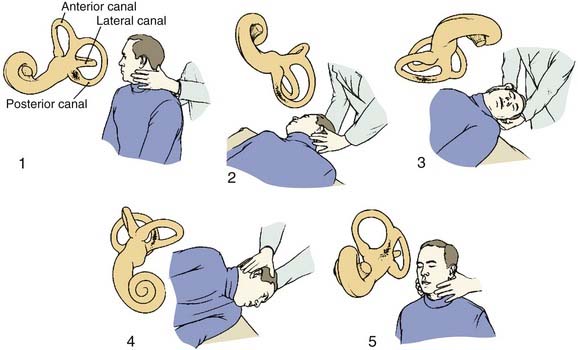
The Semont maneuver also is effective for posterior canal BPPV46 but is more difficult to perform and is less effective than the simpler, more comfortable Epley maneuver.47 The patient is moved quickly into the position that provokes the vertigo and remains in that position for 4 minutes. The patient is then turned rapidly to the opposite side, ear down, and remains in this second position before slowly sitting up.
For both Epley and Semont maneuvers, gravity is the stimulus that moves the particles within the canal, so turning the head on the body is unnecessary. En bloc movement of the head and body to the extent possible is the preferred treatment pattern. Some physicians also use a small handheld vibrator over the mastoid and claim slightly better results, although the results do not seem significantly different.48 Mastoid vibration should be avoided in patients who have had retinal detachment or who may be susceptible to such a detachment because of high myopia. Some investigators recommend having the patient sleep with the head elevated for 1 to 2 days after the maneuver, but this has not been shown to have a significant effect.49
Treatment maneuvers also have been suggested for the lateral canal variant of BPPV. In cases that involve geotropic nystagmus, lying on one side with the affected ear up for 12 hours has been reported to be effective in the vast majority of cases.50
Treatment maneuvers for superior canal BPPV should follow the same principles. The head initially should be moved into a dependent position wherein the ampulla is superior, then rotated by 180 degrees, and then brought back to its initial position. Data on treatment of superior canal BPPV have not been reported, no doubt owing to its relative rarity.51
Surgical Treatment
Singular neurectomy to treat refractory BPPV was proposed by Gacek.25 During the three decades since it was described, the procedure has been performed at least 342 times, of which 252 were by Gacek himself.52 Although the procedure generally is effective, it is technically difficult and the risk of hearing loss from the procedure may be as high as 41%.53
Posterior semicircular canal occlusion was introduced as a treatment for BPPV in 1990.54 This technique blocks the canal lumen so that it becomes unresponsive to angular acceleration. At total of 97 cases have been reported in the literature. Although the procedure was associated with brief postoperative vertigo, 94 of the 97 patients were cured of their BPPV.55 The procedure is associated with a postoperative hearing loss that usually recovers over several weeks.
Vestibular Neuritis
Vestibular neuritis typically manifests with dramatic, sudden onset of vertigo and attendant vegetative symptoms.56 Typically, the dizziness lasts for days, with gradual, definite improvement throughout the course. Balance-related complaints, particularly caused by or related to rapid head movements, may be present for months after resolution of the acute disease. Paroxysmal positional vertigo occurs subsequently in a small percentage of patients.57,58 Vestibular neuritis has been known to recur, with some patients reporting similar, usually less intense attacks for years.58 Bilateral disease has been described and must be considered in the differential diagnosis for bilateral vestibular loss. Vestibular neuritis is not associated with subjective change in hearing or with any focal neurologic complaints.21 Although historically the terms vestibular neuritis and labyrinthitis were used interchangeably, vestibular neuritis is now considered the more accurate term for cases that do not involve hearing loss. The term epidemic vertigo has also been used historically and is synonymous with vestibular neuritis; the term reflects the observation that the vertigo often is preceded by upper respiratory infection.58
A documented caloric response reduction or positive head thrust test in the direction of the involved side can be used to identify the side of involvement.56 Head thrust testing in the canal plane may have the advantage of localizing the pathology to the inferior or superior vestibular nerve.59 However, inferior vestibular nerve involvement is unusual, so VEMP testing is rarely useful.60 Lateralization also has been reported with use of magnetic resonance imaging (MRI) with gadolinium enhancement.61
Histopathologic studies of temporal bones from patients believed to have experienced this clinical entity reveals vestibular nerve degeneration with sparing of the peripheral receptor structure.58 Human pathologic studies also have demonstrated signs of more chronic inflammation in the rare case manifesting a more chronic or recurrent course of disease. A postinfectious syndrome has been suggested in these cases. Opinion holds that the superior vestibular nerve is more commonly involved in this disease than the inferior. It is postulated that this predilection may be secondary to the longer and narrower bony canal traversed by the superior nerve, making it more susceptible to compressive swelling.62 It also is possible that isolated inferior vestibular nerve neuritis is not diagnosed owing to normal horizontal head thrust and caloric testing.63
Treatment has historically been supportive and symptomatic for vertigo and related vegetative symptoms. Recently, methylprednisolone and valacyclovir have been investigated as possible therapeutic agents in a placebo-controlled, double-blind study.64 Patients given methylprednisolone experienced significantly more improvement after 1 year. Valacyclovir was not shown to have any affect. As in all acute peripheral vestibular losses, early ambulation is encouraged.
Meniere’s Disease
History
Prosper Meniere first described the symptom complex in 1861 and proposed the pathologic site to be in the labyrinth. It was Meniere along with Flourens who recognized that vertiginous symptoms could originate in the inner ear. However, hemorrhage into the inner ear, which Meniere himself believed to be the pathophysiology, has proved to be erroneous. Knapp advanced the hypothesis that hydrops was similar to ocular glaucoma,6 although this was not histologically demonstrated until 1938.12,13 Before that time, “Meniere’s disease” was used as a generic term for any peripheral vertigo. Understanding of Meniere’s disease has advanced considerably since these initial descriptions, yet the cause of the underlying hydrops remains elusive and controversial despite seventy years of active research.
Incidence
Wide variation exists in the published rates for incidence of Meniere’s disease. These range from 17 per 100,000 in the Japanese population65 to a high of 513 per 100,000 in the population of southern Finland,66 with several studies reporting intermediate values. Some of the variation may be explained by the diagnostic criteria used and by access to health care in a population. The disease seems to be more prevalent among whites,67 with a male-to-female ratio of approximately 1. The peak age at onset is in the fourth and fifth decades of life, although presentation can be at almost any age.
The frequency of bilateral disease is unclear, and the incidence in published reports is from 2% to 78%.68 The rate depends on the duration of follow-up and the diagnostic criteria. The studies at the extreme ends of this range were from before 1980, when standardized diagnostic criteria were not in use. The true incidence probably is in the range of 19% to 24%.67,68 Symptoms of bilateral disease may appear many years or decades after onset of the unilateral symptoms.69
Familial occurrence of Meniere’s disease has been reported in 10% to 20% of cases.70 An autosomal dominant mode of inheritance has been suggested,71 although the mode of inheritance can be variable. Migraine is strongly associated with Meniere’s disease in familial cases.72 The incidence is elevated in persons with specific major histocompatability complexes (MHCs). Human leukocyte antigens (HLA) B8/DR3 and Cw7 have been associated with Meniere’s disease.73,74 The etiology of the disease in persons with these HLAs may be autoimmune.75
Pathogenesis
The pathologic basis thought to underscore these findings is a distortion of the membranous labyrinth. The hallmark of this distortion is endolymphatic hydrops.12 This condition reflects the changes in the anatomy of the membranous labyrinth as a consequence of the overaccumulation of endolymph. Such changes occur at the expense of the perilymphatic space (Fig. 165-3).
Traditional thought has held that endolymph, which is produced by the stria vascularis in the cochlea and by the dark cells in the vestibular labyrinth, circulates in both a radial and a longitudinal fashion.76 In the case of hydrops, the underlying pathophysiology is controversial, but inadequate absorption of endolymph by the endolymphatic sac is the prevalent theory.77 The endolymphatic duct may act as a valve to regulate endolymph homeostasis.78 Temporal bone histologic equivalents of hydrops have been produced in animals as a consequence of disruption of the endolymphatic sac, thereby supporting the above theory of pathogenesis.79
Histopathologic studies of the human sac in the hydrops patient remain controversial. Some investigators have reported perisaccular fibrosis80 and decreased endolymphatic duct size.81 However, the underlying cause of these histologic findings is not certain.
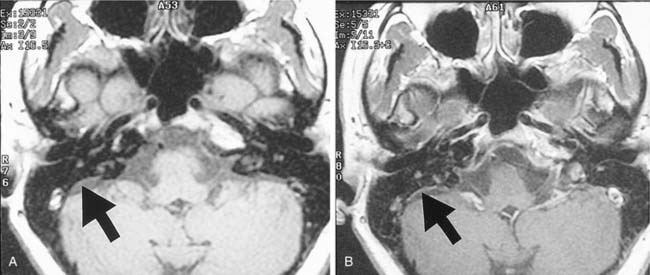
Imaging studies in patients with Meniere’s disease also identify abnormalities of the endolymphatic drainage system. Such studies suggest hypoplasia of the endolymphatic sac and duct, reflected in the decreased visualization of the vestibular aqueduct and reduction in periaqueductal pneumatization on computed tomography (CT) imaging.82 Persons with Meniere’s disease have significantly smaller and shorter endolymph drainage systems, as measured by the distance between the posterior semicircular canal and the posterior fossa on magnetic resonance images.83 These anatomic variations are apparent by the age of 3 years and may predispose affected persons to later development of Meniere’s disease. Enhancement of the endolymphatic sac84 (Fig. 165-4) and perilymphatic space85 has been demonstrated on MRI after gadolinium enhancement. However, significant individual variation has been observed, so imaging studies are neither diagnostic or predictive for Meniere’s disease. The main role of imaging in the diagnosis of Meniere’s disease is to exclude other possible causes of dizziness and unilateral hearing loss, such as a vestibular schwannoma.
Endolymphatic hydrops has been uniformly observed in the temporal bones from persons with Meniere’s disease. However not all patients found to have hydrops, had a history of Meniere’s disease.86,87 Hydrops also is a postmortem finding after labyrinthitis, otitis media, head trauma, mumps, and meningitis. Asymptomatic hydrops also has been described.88
Ruptures in the membranous labyrinth of the patient with Meniere’s disease are thought to be significant to the pathophysiology of the disorder. Membranous ruptures have been found in nearly all parts of the inner ear. Healed scars, presumably after rupture, also have been identified.89,90 Their presence has supported one of the more prominent theories of the pathogenesis of Meniere’s disease. Schuknecht postulated that ruptures in the membranous labyrinth allow leakage of the potassium-rich endolymph into the perilymph, bathing the eighth cranial nerve and lateral sides of the hair cells.91 High concentrations of extracellular potassium depolarize the nerve cells, causing their acute inactivation. The result is a decrease in auditory and vestibular neuronal outflow consistent with the hearing loss and features of acute vestibular paralysis seen in a typical Meniere’s attack. Healing of the membranes is presumed to allow restitution of the normal chemical milieu, with termination of the attack and improvement in vestibular and auditory function. The chronic deterioration in inner ear function presumably is the effect of repeated exposure to the effects of the potassium. This theory remains somewhat controversial, because some investigators have suggested that these ruptures rarely occur and do not adequately explain the observed symptoms.92
Etiology
The triad of hearing loss, tinnitus, and vertigo constitutes Meniere’s syndrome. If the cause is unknown, it is defined as Meniere’s disease.93 However, if a disease entity that is known to cause endolymphatic hydrops is associated with the syndrome, the diagnosis is one of secondary endolymphatic hydrops (i.e., otosclerotic foci causing mechanical endolymphatic blockage94). Obstruction of the endolymphatic duct is the basis for development of hydrops in experimental animals. This is accomplished by any lesion that can produce failure of duct function, including mechanical blockage, chemical fibrosis, viral inoculation, immunologically induced inflammation, and ischemia.95 However, these animal models cannot be interpreted to explain the actual cause of the human disease. These models also do not completely reproduce the clinical and pathologic entity experienced by humans.
Antibodies directed against normal inner ear elements have been suggested. Patients with Meniere’s disease have an increased incidence of specific types of human leukocyte antigens.70,96 However, most autoimmune processes, including those that affect the ear, such as Cogan’s syndrome, have histopathologic findings including infiltration of white blood cells and cellular destruction. If an autoimmune mechanism is responsible, it would have to have a more indolent course or be intermittent and absent at the time of tissue sampling.
Viral infection also has been a suggested pathomechanism for Meniere’s disease.89 The observed occurrence of symptomatic hydrops many years after unexplained deafness, so-called delayed endolymphatic hydrops, suggests that subclinical viral infection could cause hydrops many decades later.97 However, no virus has been conclusively identified, and comparison of patients with Meniere’s disease with control subjects has demonstrated no difference in response to herpes simplex virus.98
Ischemia of the endolymphatic sac or inner ear has also been proposed as a underlying mechanism of Meniere’s disease.99 Such a common vascular mechanism may link migraine and Meniere’s disease.72,100,101
Numerous factors have been implicated as causative for Meniere’s disease. Although this lack of specificity in part reflects the current lack of understanding, it also suggests that Meniere’s disease may be multifactorial or may represent the common end point of a variety of injuries or anatomic variables. A number of processes seem to be associated with the development of hydrops (e.g., trauma, acute otitis media, labyrinthitis, congenital inner ear deformity, idiopathic processes), but these are not always associated with the development of symptoms.86 It is possible that Meniere’s disease is precipitated by a variety of events such as autoimmune, viral, traumatic, or vascular or ischemic and may even be the result of congenital anatomic and molecular variations that may act as triggers for the later development of symptomatic hydrops (see Chapters 149 and 153).
Diagnosis
No single test is available to make the diagnosis of Meniere’s disease. Rather, the most important aspect of the diagnostic process is a complete history, including a detailed description of the pattern of disease presentation, supported by quantitative testing. The most recent definition of the disease has been established by the Committee on Hearing and Equilibrium of the American Academy of Otolaryngology–Head and Neck Surgery (AAO-HNS) and is summarized in Table 165-1.102
Table 165-1 AAO-HNS Criteria for Meniere’s Disease Diagnosis
| Major Symptoms |
| Vertigo |
| Deafness |
| Tinnitus |
| Diagnosis |
| Possible Meniere’s disease |
• Sensorineural hearing loss, fluctuating or fixed, with dysequilibrium, but without definite episodes |
| Probable Meniere’s disease |
| Definite Meniere’s disease |
| Certain Meniere’s disease |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


