Periocular Mesenchyme: Neural Crest and Mesodermal Interactions
Christina Wahl
Periocular tissues consist of oculomotor muscles, connective tissues, glands, blood vessels, and nerves of the orbit within which the eye is supported and suspended. Although the progenitors of these tissues and their embryonic histories are distinct from those of the eye, they are nevertheless intimately integrated with ocular development. Most periocular tissues arise from three basic embryonic cell types—neurepithelium, neural crest, and mesoderm. In addition, ectoderm contributes the epidermis of the eyelids.
The objective of this chapter is to discuss what is known about the early development and morphogenesis of the periocular area and especially to consider the various proposed mechanisms of spatial assembly, patterning, and differentiation of its principal contributors, the neural crest and mesoderm. Much of this information was gathered from avian, amphibian, fly, frog, mouse, and zebra fish species. Comparisons of growth patterns among them show enough similarity that we can, with confidence, extrapolate to the human embryo, for which only descriptive data exist.
To construct the head of the embryo, zygotic cells must overcome a series of developmental problems. The first major problem is establishment of the body axis; the second is formation of the mesoderm; the third is differentiation of the neural crest; and the fourth is integration and interaction of the mesoderm and neural crest. Embryonic cells must grow, divide, and move rapidly—activities that cannot occur simultaneously due to limitations imposed by the cell cycle. Initially, local environmental cues serve to coordinate the activity of whole populations of cells and are accomplished through progressive changes in spatial organization and induction (signaling between neighboring cell populations). Later, paracrine signaling mechanisms become more precise, allowing individual cells to respond to specific, spatially restricted signals. These interactions produce “cell lineages” through gradually established, selective gene silencing patterns that differentiate the cells, separating mesoderm into the muscles and vascular endothelium, and neural crest into the cornea, nerve, and connective tissues of the orbit.
During the past 10 years, the major advances in our understanding of how the periocular environment is assembled have come from molecular cell biology and genetics. This chapter attempts to integrate these new advances with our established understanding of embryonic anatomy and spatial patterning processes.
What Regulates Differentiation within the Nucleus?
Since the human genome was sequenced in 2003, the next big challenge has been to determine how it is functionally organized, and how gene expression and silencing are controlled.1 Such controls are mediated through the presence of DNA-binding transcription factors, as well as through epigenetic mechanisms like DNA methylation. Because zygotic cells are pluripotent, they can readily alter their gene expression patterns according to the cellular environment in which they find themselves. This is possible because much of the genome is available for transcription in a pluripotent cell, whereas in the fully differentiated state, many genes have been permanently silenced.2
Events leading to terminal differentiation are: (a) signaling and movements between cell populations leading to (b) appropriate up- or down-regulation of transcription factors, followed by (c) heritable gene silencing. Heritable gene silencing is maintained throughout all generations in a given cell lineage, thus ensuring stable maintenance of phenotype. Signaling among pluripotent cells is effected by direct contact with matrix components or neighbor cells, and by morphogens. The “cocktail” of morphogens, matrix, and neighbors around a cell affects its behavior through up- or down-regulation of transcription factor expression. Morphogens may bind to membrane receptors, thus stimulating second messenger signaling within the cytoplasm to effect transcriptional regulation. This type of signaling requires competence in the target cell—the appropriate receptors must be present. For example, only cells displaying the Patch receptor can respond to hedgehog signaling proteins.3 Other morphogens diffuse directly into the cell to interact with nuclear receptors. An example is retinoic acid (RA), which interacts with cytosolic receptors. The RA-receptor heterodimer is a transcription complex, entering the nucleus and binding to RA-responsive DNA elements (RAREs) located in the regulatory regions of target genes.4 Thus, several transcription factors may be required to form an active transcription initiation complex that helps RNA polymerase to bind to a particular gene’s promotor. To compound this complexity, transcription factors can themselves act in either a cell-autonomous fashion, through regulation of genes influencing differentiation in the cell where they are expressed, or nonautonomously, by regulating expression of morphogens that affect surrounding cells.5
Transcription factors either may activate or repress gene transcription, and they are dynamically associated with chromatin, rapidly exchanging between regulatory elements and the nucleoplasm. This “hit-and-run” behavior, combined with the presence of molecular chaperones that promote continuous disassembly of some transcription complexes, increases a cell’s ability to respond to changes in its environment.1
Intriguingly, it is through regular activity that some genes are retained in the transcriptionally active state, whereas infrequently transcribed genes become increasingly more difficult to activate, and the least active genes are permanently silenced.2 The “use it or lose it” principle apparently applies to the differentiation of gene expression patterns in different cell lines!
Both gene expression and gene silencing have important implications during development. The silencing of genes restricts a cell lineage to a reduced number of possible phenotypes, producing a form of epigenetic memory. Of the two forms of gene silencing, Polycomb-trithorax (Pc-G/trx) occurs in worms, flies, and vertebrates, whereas CpG dinucleotide methylation is restricted to vertebrates. Histone modification leading to gene silencing also can influence gene transcription, but it is not yet clear whether these modifications are heritable.2
Fertilization is followed by a period of genome-wide demethylation, erasing the original parental imprinting patterns. Later, during the formation of the blastula, early CpG methylation activity is involved with large-scale silencing—X chromosome inactivation and genomic imprinting are examples.6
Lineage-specific gene silencing is established later, as the embryo grows and cell lines are established through induction and spatial programming. Different cell lines are effected through the spatial expression of maternal and axial patterning genes and by local induction between ectoderm, endoderm, and mesoderm. As differentiation progresses, silent genes are more likely to be methylated, and the degree of methylation determines the amount of activation signal necessary to activate expression later. Weakly methylated genes are more responsive than are heavily methylated ones. Through these genomic mechanisms, cells become differentiated. They begin to show the phenotypic characteristics associated with the endothelium, muscle, nerve, and connective tissues of the periocular environment.
Establishing the Blueprint
Because the zygote is essentially totipotent and has no endogenous “molecular map,” proper spatial organization is established in the earliest stages by maternally derived morphogens. The body axis is defined in the blastula, before the embryo exhibits any overt polarity, through morphogen signaling that leads to the establishment of a node or head organizer. Cells move rapidly around and through the node during gastrulation, defining a shallow rostrocaudal depression known as the primitive streak. Next, mesoderm forms along the primitive streak (Fig. 1.1), and it must be imbued with regionally specific properties such that, acting in concert with overlying ectoderm, it can effect the spatial organization of axial structures. Antero-posterior and dorso-ventral patterning is controlled by several families of genes, all highly conserved. For example, Hox genes produce transcription factors that contain highly conserved sequences of amino acids called homeodomains. These homeodomains consist of four α-helix segments—two that bind directly to DNA and two that are thought to control binding specificity.7 The Hox gene regulatory elements are key to developmental patterning processes. Different combinations of Hox transcription factors serve to delineate cell fates at each axial level (see reference 8 for review). The expression of Hox genes is initially confined to the neural tube caudal to the second rhombomere,9 but as development progresses, Hox expression changes, and Hox transcription factors are later involved in patterning of the eye and periocular region (discussed below).
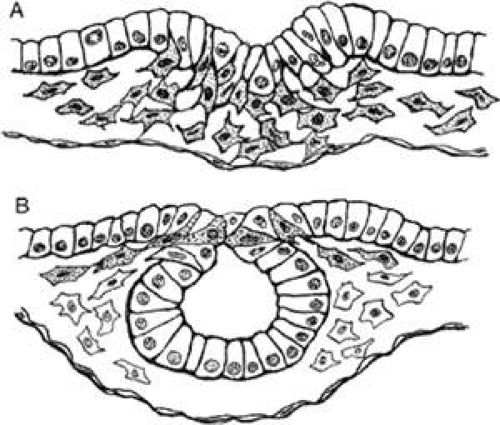 Figure 1.1 Epithelial-mesenchymal transformation. A. Mesodermal mesenchymal cells (stippled) are delaminating from the region of the primitive streak. B. Neural crest cells (stippled) are delaminating from the neural folds. |
The body axis is defined by the genesis of the node and primitive streak. The region just anterior to the node is called the prechordal plate. Mesoderm, largely responsible for dorso-ventral and antero-posterior patterning of the body axis,10,11 also underlies the future eye-forming part of the prechordal plate, or eye field, before neural tube closure. Mesoderm produced at the pre-chordal plate leads to formation of the prosencephalon and diencephalon and related head structures.
Patterning of the prechordal plate differs substantially from that of the rest of the head and trunk. Caudal to the node clear rostrocaudal organization occurs, maintained by several Hox genes. Different combinations of Hox genes control segmental identity of the hindbrain and trunk, but the prosencephalon is not part of this organization. It forms as a single, uninterrupted crescent-shaped area extending forward of the node, with no primitive streak. However, interruption of Hox gene expression in the most anterior hindbrain segments can have a direct impact on the head. For example, the Hox gene XLim 1 (and its mammalian homologue Lim 1) and the zinc-finger–containing Hox gene Krox 20 produce transcription factors that are essential to the establishment of anterior head structures. Both Lim–/– and Krox 20–/– mutants are truncated just rostral to rhombomere 3, in the metencephalon.12 Many cis-elements and regulatory factors contribute to segmental activity, as demonstrated in a study of Krox20. Krox20 controls the expression of other genes, including Hoxa2 and Hoxb2, by binding to enhancers that flank these genes. The expression of Krox20 itself is regulated in a negative feedback loop with other corepressors.13
Mesoderm is generated in the prechordal region, as elsewhere, via a process known as epithelial-mesenchymal transformation (see Fig. 1.1), through which epithelial cells lose their tight junctions and move between openings in the underlying basement membrane. Mesenchyme is a unique embryonic connective tissue that lacks tight junctions between its highly mobile cells (all other embryonic cells at early stages have extensive tight junctions), and it includes an extensive extracellular matrix (ECM). Mesenchymal cells advance by “pulling” themselves through the filaments of the ECM, a process that depends on the presence of certain cell adhesion–specific molecules in the ECM and complementary matrix adhesion–specific molecules on the cell surfaces. This has been demonstrated both in vitro and in vivo. For example, when ectodermal explants are treated with heparinase to remove glycosaminoglycans, morphogenetic movements are inhibited and no mesoderm is formed.14
Mesoderm may be restricted, as opposed to induced, through the action of bone morphogenetic proteins (BMP transcription factors), members of the transforming growth factor–β superfamily. These morphogens are important during the early fate-mapping process,15 although subsequently they too may assume different signaling roles. BMP 3 inhibits noggin and goosecoid, thus restricting a subset of mesoderm that contributes to the head and anterior trunk (dorsal mesoderm).16 Maternally derived BMP 2 promotes generalized mesoderm formation,17 whereas BMP 4 is more specific, inhibiting the production of dorsal mesoderm and promoting the differentiation of ventral structures, including the blood, pronephros, and visceral tissues.18
The formation of mesoderm from transformed ectodermal cells completes the three primary germ layers: ectoderm, mesenchymal mesoderm, and endoderm. Next, many important lineage commitments relative to the eye and periocular tissues become established. The crescent-shaped eye field arises rostral to the primitive streak. This eye field, which is initially located in the prechordal plate, must bilateralize to produce two eyes (Fig. 1.2; also see elsewhere in these volumes). When the prechordal mesoderm underlying the eye field of Xenopus embryos is removed, only one retina is formed.19 One of the most important signals involved in separation of the eye field is hedgehog (sHH).20 The sHH protein is first expressed along the body axis and the posterior margins of limb buds. On binding to the Patch receptor, sHH relieves repression of the G-protein–coupled receptor Smoothened. Smoothened acts intracellularly via Gli proteins to activate or repress target genes.21 The sHH knockout mouse phenotype is lethal, and displays incompletely fused neural tubes with poorly defined ventral midlines, severe cyclopia, an overlying proboscis, and no olfactory placode.22 In humans, sHH mutations also cause holoprosencephaly.23 Interestingly, mutations in the human Gli proteins cause hypertelorism, as in Grieg cephalopolysyndactyly.24
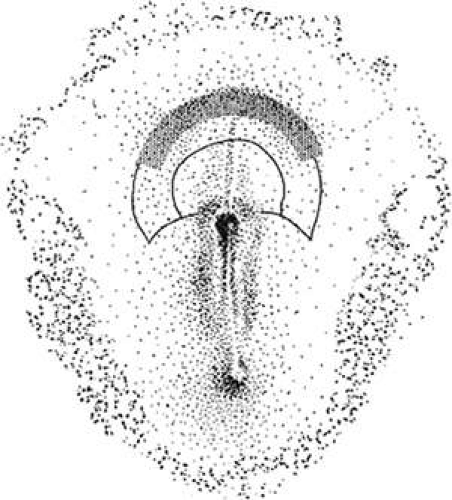 Figure 1.2 Pax-6 expression in the prechordal plate (25,20). The borders of the presumptive basal and alar lamina (inner solid line) and presumptive neurosomatic junction (outer solid line) are shown. The stippled crescent in the anterior prechordal plate represents the eye field, as recognized by Pax-6 expression. |
Several other transcription factors expressed in the eye field are essential for eye formation.25 These include ET, Rx1, Pax6, Six3, Lhx2, tll, and Otx2 (also known as Six6). A cocktail of these factors, together with Otx2, is sufficient to generate ectopic eye expression outside the vertebrate nervous system, as demonstrated in an elegant study by Zuber et al. (Fig. 1.3).25 Otx2 overrides the inhibition of ET by noggin (a BMP 4 antagonist),26 allowing the expression of the other transcription factors required to form eyes. It is possible to experimentally interfere with the expression of one of these eye field transcription factors and demonstrate the fidelity of the pathway. For example, when the Drosophila Pax-6 homologue eyeless is artificially upregulated in larvae, ectopic eyes form on the legs, antennae, wings, and elsewhere.27 This demonstrates that the general functions of many key regulatory genes have been conserved through thousands of millennia of animal evolution.
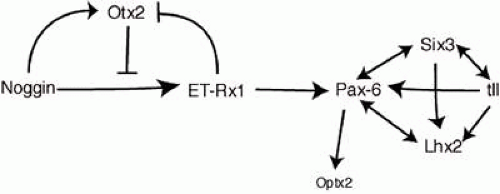 Figure 1.3 Specification of the eye by interactions among eye field transcription factors. Arrowheads indicate stimulatory effects, “T” bars indicate repressive effects. Reciprocal stimulation is shown by arrowheads at both ends of a line. (Reproduced with permission from Zuber ME, Gestri G, Viczian AS, et al. Specification of the vertebrate eye by a network of eye field transcription factors. Development 2003;130(21):5155–5167.) |
Usually, teratogenic agents are blamed for idiopathic failure of the eye field to separate. Veterinarians have long been aware that pregnant ewes grazing on fields containing specimens of the weed Veratrum give birth to cyclopic lambs if they eat the plant during the first weeks of gestation.28 The most common teratogen affecting human births is ethanol, which disrupts the prosencephalon by limiting its size; the affected child may show microphthalmia, short palpebral fissures, deficiencies of the philtral region, and a long upper lip.29 The otherwise low incidence of cyclopia in nature suggests that chemical agents with the potential to cause major prosencephalic defects generally are not encountered during the short window of development when the embryo is vulnerable.
The periocular area undergoes marked changes in tissue relations as the brain grows. As the flat neural tube expands mediolaterally, its walls form paired lateral outpockets called optic vesicles, which involute to become the optic cups (Fig. 1.4). The prechordal and paraxial mesoderm originally underlying the rostra1 neural plate expands laterally and rostrocaudally in congruence with the neural tissue until the evagination of the optic vesicles pushes adjacent mesoderm caudally and ventrally. This leaves the optic vesicle devoid of an enveloping mesenchymal layer except on its caudal (future temporal) surface. This “temporal” mesenchyme is not displaced or migratory but consists of a uniform population of mesodermal, stellate cells located in the 12- to 6-o’clock positions. The remaining periocular area is acellular, consisting solely of ECM in the space between the optic vesicle and overlying ectoderm.
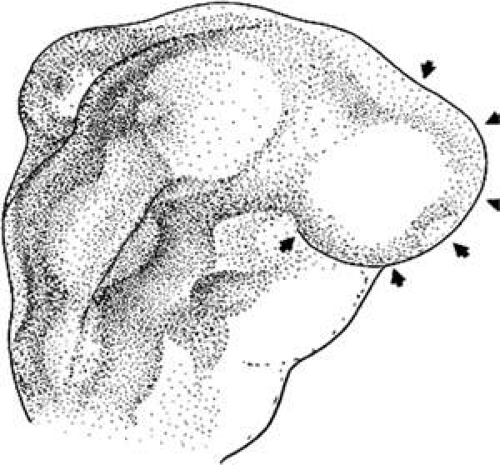 Figure 1.4 Formation of the optic vesicles as lateral bulges of the prosencephalon. This drawing is a dorsal view of the right side of an avian embryo at about 2.5 days’ development. The arrowheads indicate the superficial extent of the bulging right optic vesicle. |
The neural epithelium of the optic vesicle is now in direct apposition to the surface ectoderm along its lateral and anterior surfaces. Normal development of the eye depends on the absence of mesenchyme in these areas, as has been demonstrated by the mouse mutant extra-toes, in which invasion of mesenchymal cells along the lateral margin prevents normal formation of the lens.30 In normal animals, mesenchyme that may become trapped between the invaginating optic vesicle and thickening lens undergoes necrosis and is resorbed. In a congenitally anophthalmic mouse strain, this trapped mesenchymal population failed to undergo apoptosis, and the normal sequence of necrotic loci in the developing eye was lacking.31
The failure of the lens placode to contact the optic vesicle as a result of inappropriately placed mesenchyme results in anophthalmia among these genetically defective animals.32,33 The lens placode also plays a key role in ocular expansion, perhaps related to the generation of the vitreous. Growth of the retina is unaffected by eye size, because the retina will continue to proliferate beyond the confines of a miniaturized eye. Mesenchymal processes that lead to expansion of the globe do not control or otherwise affect the mitotic rate within the retina. Retinal convolutions may fill the eyecups of microphthalmic or anophthalmic animals, a phenomenon that can be duplicated experimentally through the use of teratogens.34
Cell Death as a Tissue-Shaping Mechanism
The observations of Veermeij-Keers and Poelman35 focus on the high degree of cell proliferation, degeneration, and differentiation associated with neural crest. These processes should certainly be taken into account in considering the overall phenomenon of crest cell migration, because facial malformations may result from errors in any one or more. A specific example of the role of cell death in crest cell migration is seen in the chick hindbrain at the level of rhombomeres 3 and 5, where no crest is observed to emerge, but cell death at the dorsal lips of the neural folds is marked. This suggests that progenitor crest cells are selectively eliminated in these areas.36
Excessive apoptosis within both cranial mesenchyme and the neural tube correlates with exencephaly and facial clefting, but no change in AP-2 expression, in mouse embryos deficient in a transcription factor produced by the proto-oncogene c-ski.37 Severe hypoplasia of skeletal muscles is also part of the ski –/– phenotype.37 The hypoplasia appears to affect muscle growth only after terminal differentiation, because upregulation of ski causes secondary myoblast proliferation.38
Primary myotubes degenerate as secondary myotubes become established, but then the secondary myotubes also undergo remodeling through local apoptotic foci.39 Clearly, abundant cell proliferation is a hallmark of development; however, as these examples illustrate, cell death is another important shaping tool at work in the embryo.
The Neural Crest: Formation of Periocular Connective Tissues
If the optic vesicle were to remain devoid of surrounding mesenchymal cells, both its development and the potential to form the array of connective tissues necessary for orbital and corneal development would be absent. It is not known why the existing mesodermal cells in early vertebrates did not simply migrate rostrally and circumscribe the globe. Instead, a new population of mesenchymal cells, the neural crest, was generated to serve this and many other functions.
The neural crest is a highly migratory cell population, it carries great differentiation potential, it exerts inductive influences on almost every tissue that it invests, and it is essential in midfacial and orbital development. Students of comparative neuroanatomy hear the neural crest defined as “the single greatest innovation separating vertebrates from invertebrates.” The importance of this embryonic tissue extends beyond the fact that it forms all autonomic postganglionic neurons, the enteric nervous system, the outflow tract of the heart, and melanophores. In the formation of the peripheral glia, or Schwann cells, the neural crest furnishes a way to speed conduction velocities along axons. Saltatory conduction, arguably more so than any other feature, explains why the world’s largest animals are vertebrates. Much of the vertebrate head is neuroectodermal in origin, because most of its bones and all its fibroblast-derived connective tissues, dermis, and perivascular smooth muscles arise from neural crest. Only the endothelium, glands, and muscles of the orbit are not crest derivatives. Thus, the formation and migration of the neural crest into the periorbital region signals the beginning of final tissue integration and assembly, as required for scleral formation, development of the accommodative mechanism of the eye, and spatial patterning of the eye muscles.
The neural crest is formed around the time of closure of the dorsal lips of the neural tube, during a second period of epithelial–mesenchymal transformation limited to the angle in the epithelium between dorsal ectoderm and neural tube (Fig. 1.5). A transcription factor that is inducible by retinoic acid, AP-2, is expressed by crest cells and neurepithelial cells at the neural folds. AP-2 null mice display cranio-abdominoschisis and severe dysmorphogenesis of the face, skull, sensory organs, and crest-derived cranial ganglia.40,41 AP-2 is not required for neural crest migration, but it does appear to be necessary for the regulation of neural tube closure, and its absence causes abnormalities in crest-derived structures.41
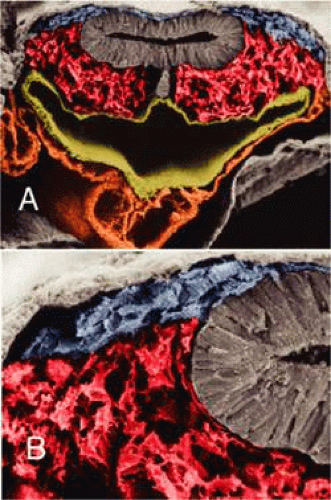 Figure 1.5 Scanning electron micrographs of embryos colorized to indicate mesoderm (red), ectoderm and neural tube (gray), pharyngeal endoderm (tan), and neural crest (blue). In A, the neural crest is seen in the angle between dorsal ectoderm and neural tube. At this time, the neural crest is a distinct population that has not mingled with the paraxial mesoderm, as seen in B. |
Cephalic crest cells start their migration at the four-somite stage in humans42 and at the five-somite stage in mouse43 and avian embryos.44,45,46 They do not arrive in the periocular region until the optic vesicle has formed. The crest population expands while the optic vesicles enlarge, and the optic stalks begin to constrict. This produces a space between the dorsal surface of each stalk and the overlying surface ectoderm that is soon occupied by neural crest cells; some of these are diencephalic in origin, but most have moved rostrally from the region of the metencephalon.
Like mesodermal mesenchyme, movements of neural crest cells depend on molecular interactions with the ECM. In the chick embryo, neural crest cell movements are disrupted if HNK-1, an antibody to a carbohydrate moiety involved in cell adhesion, is injected into the ECM along migration pathways.47 In albino axolotls, it has been shown that fibronectin, tenascin, collagens I and VI, and a chick aggrecan stimulate the migration of crest cells, although in vitro, the aggrecan molecule inhibits cell migration.48 (This latter result is a dramatic reminder that cell responses to specific molecules within three-dimensional, intact organisms may differ substantially from behaviors in culture.) The molecular structure of the ECM and epithelial tissue boundaries thus delineate the migration routes of the neural crest cell.49,50,51,52 Continual modification of the ECM by the mesenchyme provides temporal control over crest cell migration as substrate qualities are altered.53,54,55 Therefore, correct morphogenesis at this time depends first on the initial spatial guidance of crest cells by the mesenchyme; second, on temporal control of crest cell migration by the ECM; and third, on the behavior of the crest cells themselves en route to their final destinations.
In mammals, neural tube closure begins in the future occipital or cranial cervical region (at the level of somites two through seven in humans and four through six in rodents) and then proceeds cranially and caudally. In humans, the mesencephalic crest population that contributes to the periocular area emerges during the four- to 12-somite period.42 A second group of crest cells is reported to emerge from the optic vesicle and sulcus (or diencephalic neural folds) during the 13- to 20-somite period,42 although this population has not been experimentally traced in other species (Fig. 1.6).
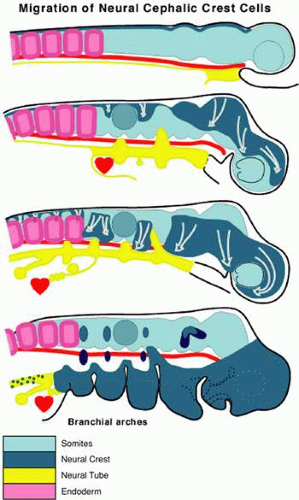 Figure 1.6 Diagram of cephalic neural crest cell migrations. Successively older embryos are indicated from top to bottom. Arrows indicate the general direction of crest cell migration. The small dark ovals in the oldest embryo (bottom drawing) represent placodes; the trigeminal ganglion is shown along the caudal mesencephalon. |
Although cranial flexure precedes neural crest migration in many mammals, including the human,42,56 in birds and some reptiles the head of the embryo is still in a flat plane as the crest cells leave the neuroectoderm. Crest cells begin to migrate en masse laterally from atop the middle of the mesencephalon. Immediately ahead of the moving front of crest cells, the ECM expands, creating an acellular space filled with hydrated glycosaminoglycans, laminin, and fibronectin. The neural crest population moves laterally through this acellular matrix, then proceeds ventrally, following the body’s contours. The first cranial (mesencephalic) flexure is a ventral folding at the midbrain level that brings the forebrain and paired eyes ventrally and caudally more than 90 degrees with respect to the axis of the trunk, to a position just rostral to the floor of the mesencephalon. Later, the second (pontine) flexure, which is a dorsal folding at the hindbrain level, elevates the eyes and forebrain, bringing them forward about 30 degrees with respect to the axis of the trunk. In the human, the midbrain cephalic flexure occurs early, beginning at the one- to three-somite stage, forming a right angle between the long axis of the forebrain and that of the hindbrain.57 Embryos at this stage are approximately 20 days old. By the 29th day, the pontine flexure is evident. During this interval, the eyes have changed considerably. The vesicles have become optic cups, the lens placode has formed a lens vesicle, the pigment epithelium has begun to produce melanin, eye muscle precursor populations are evident in the periocular area, and the eye has more than doubled in size (the actual proportion of growth varies among species). Expansion of the optic vesicle coincident with cranial flexure also brings about changes in the relations of mesenchymal cells to each other, including new contributions of mesenchymal cells from caudal sites of origin along the hindbrain. The neural crest cells forming the remainder of the dorsally situated population continue to migrate along surfaces of the optic cups (Fig. 1.7). It is important to recognize that the genesis of the neural crest, whether at open or closed neural folds, and whether early or later relative to cranial flexures, does not affect the fundamental relationships between the sites of neural crest origin and the terminal location of neural crest or mesodermal populations.46,49,58,59,60 At this stage, Hox dlx genes are expressed in the periocular region.7
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


