Chapter 45 Pediatric retinal degeneration in systemic inherited diseases
Children affected by retinal degeneration associated with extraocular manifestations define the inherited systemic retinitis pigmentosa (RP) syndromes (retinal dystrophies related to inborn errors of metabolism are covered in Chapter 62) (Tables 45.1 and 45.2). The gene or the group of genes involved in the syndrome belong to a specific biologic network whose mutations lead to retinal degeneration associated with manifestations in other organs defined by the specific biologic role of the protein. Retinal degeneration in these syndromes illustrates the biologic complexity of the retina. The photoreceptor cell is remarkably sensitive to alterations in many biologic pathways but the clinical presentation is often indistinguishable between syndromes with the classical features of retinal degeneration: night-blindness, visual field constriction, and reduced visual acuity. In the early phases of the retinal degeneration, the fundus may appear normal although the electroretinogram (ERG) is altered. Later, the fundus will denote the usual aspects observed in retinal degeneration: pigment mottling, pigment migration with spicules, optic disk pallor, narrowed vessels, and macular changes which are prominent and inaugural in the cone–rod dystrophies.
Table 45.1 Main extraocular features found and the corresponding systemic retinitis pigmentosa syndrome
| Extraocular clinical feature associated with RP in a child | Key feature found in systemic syndrome | |
|---|---|---|
| Stature | Nanism | Cockayne’s syndrome (cachectic), spondylometaphyseal dysplasia or spondyloepiphyseal dysplasia associated with cone dystrophy |
| Obesity | Bardet-Biedl syndrome (BBS) Alström’s syndrome, Cohen’s syndrome (truncal), MORM (mental retardation, truncal obesity, retinal dystrophy, and micropenis) | |
| Skeletal abnormality | Polydactyly | BBS, Joubert’s syndrome, Jeune’s syndrome |
| Short ribs with abnormal thorax | Jeune’s syndrome | |
| Various abnormal skeletal development | Spondylometaphyseal dysplasia or spondyloepiphyseal dysplasia | |
| Deafness | Deafness | Usher’s syndrome (variable severity according to types), Alström’s syndrome (progressive starting in childhood) |
| CNS malformations | Cerebellar agenesis (molar tooth) | Joubert’s syndrome |
| Kidney dysfunction | Polyuria-polydypsia Kidney failure | BBS, Alström’s syndrome, Jeune’s syndrome, Senior-Loken syndrome, Joubert’s syndrome |
| Endocrine dysfunction | Type 2 juvenile | Alström’s syndrome |
| Pituitary deficiency | Oliver-McFarlane | |
| Cardiomyopathy | Alström’s syndrome | |
| Cognitive impairment | Classical: Cohen’s syndrome, Joubert’s syndrome Occasional: BBS, Alström’s | |
| Teeth | Amelogenesis imperfecta | Jalili’s syndrome |
| Prominent incisors | Cohen’s syndrome | |
| Skin/hair | UV sensitivity | Cockayne’s syndrome |
| Alopecia – early hair loss | Hypotrichosis with juvenile macular dystrophy; EEM (ectodermal dysplasia, ectrodactyly, macular dystrophy) syndrome | |
| Long eye lashes | Oliver-McFarlane | |
During the last two decades, a considerable number of genes responsible for these conditions have been identified. This field will continue to greatly benefit from new genome exploration especially high-throughput sequencing.1–5 Molecular diagnosis and genetic counseling are constantly improving especially for those syndromes that are highly heterogeneous. The classical syndromes illustrating this group of diseases are described, but a growing number of overlapping phenotypes are revealed as molecular investigations improve. Moreover, different mutations in the same gene may lead to different syndromes of the same clinical spectrum (see section on the ciliopathies). Major clinical variability in each syndrome has been described. For some genes, specific mutations may lead to isolated features such as isolated RP (e.g. Usher’s syndrome gene USH2A or Bardet-Biedl gene BBS8 and BBS3).6–8
Usher’s syndrome: a deaf child who loses vision
Usher’s syndrome (USH) is the association of sensorineural deafness with progressive retinal degeneration. Usher’s syndrome occurs in 1 of every 10 deaf children (5% of all cases of congenital deafness). It is the most frequently inherited syndrome with deafness and is the most common syndrome among the deaf–blind.9–11 The syndrome is subdividied into three groups: Usher’s type 1 (USH1), type 2 (USH2), and type 3 (USH3). USH1 and 2 are usually diagnosed in early or late childhood, respectively, but may be diagnosed later in life as the visual impairment may be overlooked in a deaf child. Vestibular dysfunction is classically associated with USH1. Each type of Usher’s syndrome is autosomal recessive but genetically heterogeneous. For each type, a number of genes have been identified12–16 (Table 45.1). Molecular studies have shown clinical overlap between the classical types USH1 and USH2.17 Some Usher’s genes can be involved in either isolated deafness or isolated RP (USH2A).6,16
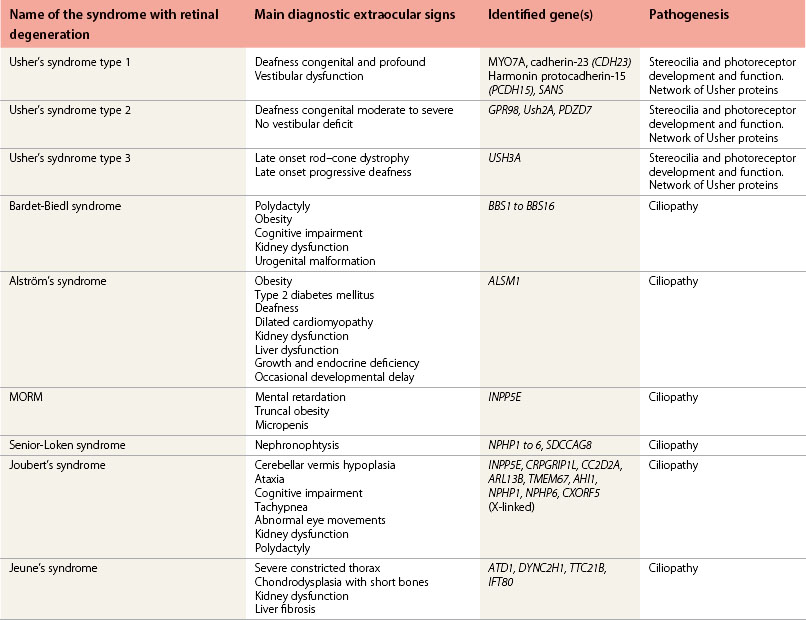
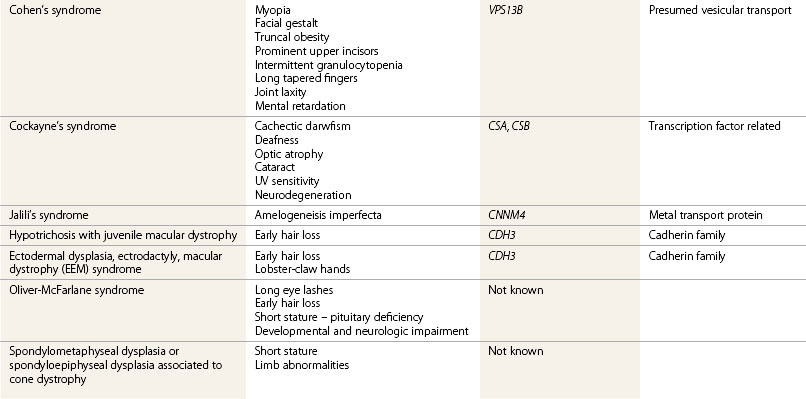
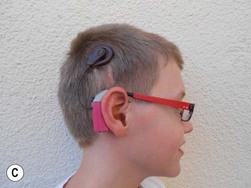
Cochlear implantation is highly recommended for these children (especially in USH1). No therapy is yet available for the retinal degeneration18,19 (Fig. 45.1A–C).
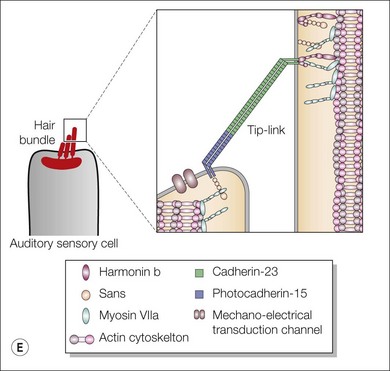
(Fig. 45.1E courtesy of Professor Christine Petit, Institut Pasteur, Inserm, UPMC, Collège de France, Paris, France.)
Usher’s syndrome type 1: the most severe form
The child with USH1 is affected with congenital severe to profound sensorineural hearing loss associated with delayed sitting and walking due to abnormal vestibular function. Retinal degeneration occurs early in childhood. As early as 2 or 3 years old, the ERG can confirm the diagnosis of retinal degeneration although the fundus appears to be normal.20,21 Five causative genes are known and mutated in various proportions according to the studied population: MYO7A (recognized to be the main USH1 gene mutated in half of the cases and also responsible for non-syndromic deafness), USH1C, CDH23, PCDH15, and USH1G.14,16 Although always severe, the course of disease may vary according to the type of mutation.22
Usher’s syndrome type 2: later onset retinal degeneration in a deaf child
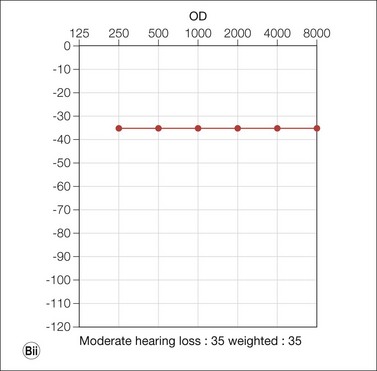
Congenital mild to severe sensorineural stable hearing loss with major impairment in high frequencies and normal vestibular function are the main characteristics of USH2. The retinal degeneration occurs later than in USH1, usually during puberty or after. USH2 and USH3 are not usually symptomatic in the first decade and initially have a normal fundus appearance. Many patients retain good visual acuity in spite of constricted visual fields. The ERG may initially be normal; however, ERG changes may be detected in presymptomatic young children. During the second decade, night-blindness and loss of peripheral vision become evident and inexorably progress (Fig. 45.1D). Three genes have been identified: USH2A (Usherin found also mutated in isolated RP), GPR98, and DFNB31.14,16
Usher’s syndrome type 3: the later onset, variable form
The USH3A gene has been identified in particular in Askenazi Jews and Finns. USH3 has also been found mutated in patients with USH1 and USH2.14,16
Pathogenesis of Usher’s syndromes
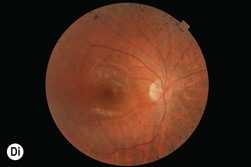
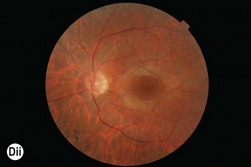
Progress has been made in understanding the pathogenesis of this disease affecting the inner ear stereocilia and photoreceptor cells of the retina.23–26 The USH1 gene products form a network, the Usher’s interactome, which plays a key role in the early hair bundle development. They are also key components of the mechanoelectrical transduction machinery necessary for hearing. In the retina, the Usher interactome acts at the level of the ciliary/periciliary region of the connecting cilia of the photoreceptor cells. It is important in the transport of proteins between the outer and the inner segments.27 Alteration of the USH proteins lead to early dysfunction of the inner ear and of the retina simultaneously (Fig. 45.1E).
The ciliopathies: a novel systemic retinal dystrophies group
A single non-motile cilium (“primary cilium”) is present in almost every vertebrate cell; motile cilia are present only in specific organs such as the respiratory or the reproductive system (Fig. 45.2A). The primary cilium is the central “antenna” of the cell allowing transduction of sensorial information from the extracellular environment to the cell. The photoreceptor cell has a connecting cilium and is, therefore, a ciliated cell.
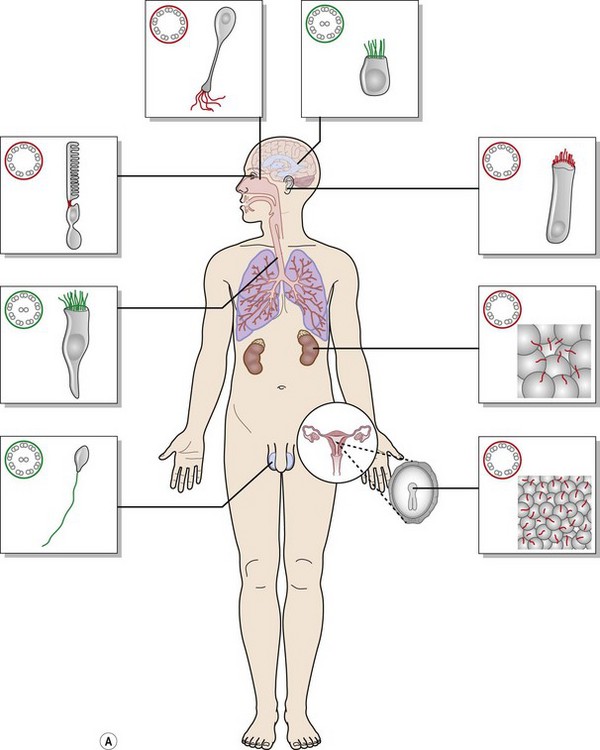
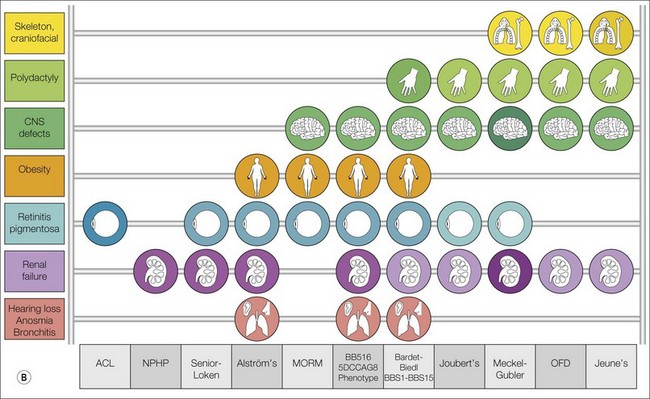
Ciliopathies are rare genetic disorders characterized by primary cilium dysfunction, frequently affecting photoreceptors and causing retinal degeneration either as an isolated condition (e.g. Leber’s congenital amaurosis with CEP290 mutations) or as a ciliopathy syndrome affecting more than one organ system.28–30 Ciliopathies involve many target organs that have in common an important role of the primary cilia. Overlapping phenotypes have been reported, as well as high clinical variability on the number of affected organs among patients with the same syndrome. The most common ciliopathies are schematically represented in Fig. 45.2B.
Molecular investigations have revealed major genetic heterogeneity for each syndrome and also allelic variability (different mutations in the same gene may lead to different syndromes). For instance, CEP290-NPHP6 truncating mutations have been identified in Joubert’s syndrome and hypomorphic mutations in Leber’s congenital amaurosis (LCA). Moreover, CEP290-NPHP6 is mutated in several distinct disorders (nephronophthisis, Senior-Loken syndrome, Joubert’s syndrome, BBS and the lethal Meckel syndrome).31
Supplementary genetic factors such as a third mutated allele in a BBS gene (known as oligogenism – a third mutation occurring in addition to the two main mutations found in recessive inheritance) or other genetic modifiers can influence the phenotype.32,33
Bardet-Biedl syndrome
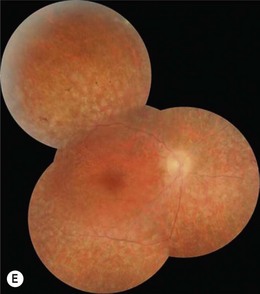
The cardinal features of the BBS syndrome are early onset retinal degeneration, obesity, polydactyly, renal failure, hypogonadism, and cognitive impairment (Fig. 45.3A–D). Secondary features may include anosmia, diabetes, cardiac anomalies, hepatic fibrosis, brachydactyly, and Hirschprung’s disease.34,35 The retinal dystrophy is early in onset leading to severe visual handicap before adulthood. Profound ERG abnormalities can be detected as early as 3 years old. Legal blindness usually results before the second decade of life.36–42 Retinal dystrophies in BBS are mainly a rod–cone dystrophy or a cone–rod dystrophy,34 usually classified as a global retinal degeneration because both rods and cones are affected29 (Fig. 45.3E).
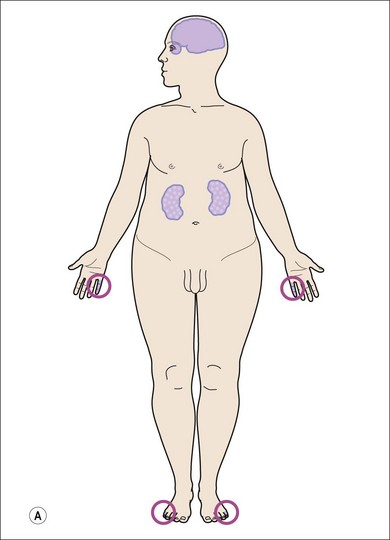
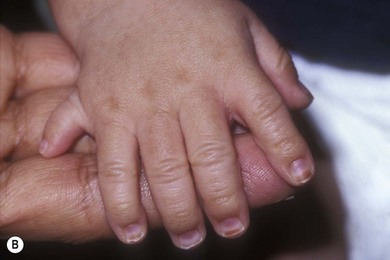
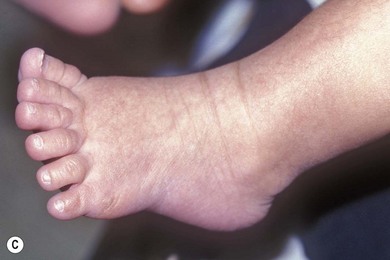

Obesity is the second major feature, present in 72–96% of BBS patients, usually beginning in early childhood and worsening with age. The origin of obesity is both central (the hypothalamic eating control) and peripheral (the adipose tissue).43–45 Limb anomalies are found in almost 95% of BBS patients, usually a postaxial polydactyly (69%). Other limb malformations such as brachydactyly or syndactyly are frequently reported for both hands and feet and have a diagnostic value. Abnormalities of the genitalia are common: hypogonadism in males or vaginal atresia in females. Rarely, there is hydrometrocolpos, a neonatal vaginal malformation leading to a massive abdominal tumor.46,47 Renal dysfunction can occur in late childhood and may lead to kidney failure.48
Neuropsychiatric symptoms can include developmental delay, mental retardation, learning difficulties, speech deficit, and behavioral problems.34 Intellectual function ranges from severe mental retardation (29%) through weak intelligence and average intelligence (29%). Slow ideation and hyperemotive status are common.
BBS is an autosomal recessive heterogeneous condition with 16 genes identified accounting for about 85% of cases.49 All the genes have been related to cilium biogenesis and/or function. BBS1 and BBS10 are the two most common, each accounting for about 20% of the cases. Implication of the other BBS genes ranges from a unique family to a few percentage of mutated families. The classical autosomal recessive inheritance model has been challenged by molecular and functional investigations with the oligogenic model and the effect of additional genetic modulators on the phenotype.33
Alström’s syndrome
Alström’s syndrome (ALMS) manifestations include RP in early childhood, hearing impairment, and metabolic defects leading to hyperinsulinemia and type 2 diabetes mellitus and obesity in childhood. Polydactyly does not occur. Dilated cardiomyopathy, often fatal, in infancy or later in life and renal dysfunction are reported in half of ALMS cases.50 Developmental or motor delays occur, but most children have normal intelligence.
The first manifestation of ALMS is severe early onset cone–rod retinal dystrophy, sometimes mimicking Leber’s congenital amaurosis with very early visual impairment, photophobia, and nystagmus (Fig. 45.4). Children usually become truncally obese during their first year. Sensorineural hearing loss presents in the first decade in up to 70%; it may progress to the moderately severe range (40–70 db) by the end of the first to second decade. Insulin resistant type 2 diabetes mellitus often presents in the second decade and is accompanied by acanthosis nigricans (pigmentation mostly in body folds). Other endocrine and metabolic abnormalities include hypothyroidism, diabetes insipidus, growth hormone deficiency, hyperuricemia, hyperlipidemia, hypothyroidism, and hypogonadotrophic hypogonadism. Hepatic and renal dysfunction can be present in the second decade. This syndrome requires a multidisciplinary follow-up to detect complications once the diagnosis is confirmed.50,51
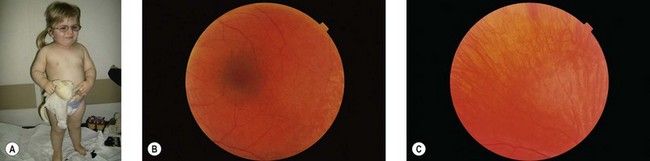
A single gene is involved in all the cases, ALMS1.52,53 ALMS1 protein is found at the base of cilia.54
MORM syndrome
MORM syndrome (mental retardation, truncal obesity, retinal dystrophy, and micropenis) is very rare.55 A congenital non-progressive retinal dystrophy with poor night vision occurs within the first year of life with reduced visual acuity by 3 years that remains stable thereafter. The identified gene, INPP5E (inositol polyphosphate-5-phosphatase), classifies this syndrome as a ciliopathy. It is related to Joubert’s syndrome.56
Senior-Loken syndrome
Senior-Loken syndrome (SLS) combines nephronophthisis (NPH) and retinal degeneration. NPH, the most common cause of inherited renal failure in childhood, is characterized by initial normal kidney size that will eventually shrink, tubulo-interstitial nephritis, and a loss of corticomedullary differentiation leading to cyst formation.30,57,58 The first symptoms are polyuria and polydipsia caused by a urinary concentration defect. The end-stage of the renal failure is variable – infantile, juvenile, or adolescent. The occurrence of the retinal dystrophy is higher in the juvenile form of NPH. The early retinal dystrophy usually occurs years before the kidney involvement is detected. The retinal dystrophy may be very early, mimicking isolated LCA or may have a later onset. Thus, clinical diagnosis of LCA should lead to clinical and molecular testing for LCA-SLS associated genes and, according to the results, annual kidney follow-up may be indicated.
Eleven genes (NPHP1 to NPHP11) are known to be involved in NPH, and for all of them, except NPHP7, cases of SLS associated with RP have been reported.30,58–61 Mutations in NPHP562 and NPHP663 are more often linked to severe and early RP. A milder retinal phenotype is observed with mutations in other NPHP genes.



