Ophthalmic Management of Craniofacial Syndromes
James A. Katowitz
Richard W. Hertle
Jeffrey B. Goldstein
Congenital and developmental anomalies of the face and cranium often present challenging diagnostic and reconstructive problems that are best handled by a team of specialists. A craniofacial team should include representatives from plastic surgery, ophthalmology, oral surgery, orthodontics, otolaryngology, neurosurgery, genetics, psychiatry, social work, and anesthesia. Each team member contributes specialized skills that enhance the diagnostic evaluation and clinical management of these patients. The relationship between the craniofacial team and each patient and family may continue for years, particularly when the patient requires multiple surgical procedures. Long-range planning must be an integral part of the team’s activities. The entire family also may be involved in the evaluation of potential hereditary diagnoses and genetic counseling.
The ophthalmologist has a unique role as both a diagnostician and surgeon. Since orbital structures represent a bridge between the face and cranium, ocular and adnexal abnormalities are frequent components of craniofacial anomalies. Subspecialists in oculoplastics, strabismus, and vision development assist in the evaluation of visual, orbital, adnexal, and motility problems, which are often present in these patients. The ophthalmologist’s responsibilities are, first, to preserve visual and protective adnexal functions and, second, to improve cosmesis. When a major deformity is recognized, interaction of the ophthalmologist and other team members is required to formulate an appropriate treatment plan.
The pediatrician or obstetrician usually is the first to recognize craniofacial abnormalities. In subtle deformities, however, the parents may be the first to recognize that something is different. The patient then is referred to the craniofacial team for more extensive evaluation. In the initial consultation, the ophthalmologist evaluates the patient for potential vision-threatening disorders and documents structural defects of the globe, as well as any orbital or adnexal deformities. Each detected anomaly is assigned a priority treatment based on its functional or cosmetic nature.
If there are no disorders that need ophthalmic treatment, then further ophthalmic evaluation depends on the timing of craniofacial surgery. If no immediate surgery is required, then the patient is seen again in 4 to 6 months as part of the craniofacial team follow-up survey. If the patient requires craniofacial surgery and early ophthalmic intervention also is necessary, ophthalmic procedures may be performed under the same anesthetic. If there are no postoperative ocular complications resulting from the craniofacial surgery, then routine follow-up is suggested 6 to 8 weeks later.
The ophthalmic surgeon’s primary responsibility involves reconstruction of the lids, canthi, nasolacrimal system, ocular surface, globe, extraocular muscles, and orbital walls. Midfacial advancement, orbital realignment, cranial vault reshaping, and major cleft repair usually are performed by other team members.
SPECIFIC SYNDROMES
The number of published classification systems describing craniofacial anomalies attests to the variety, overlapping features, and variable severity with which craniofacial patients may present.1,2,3,4,5,6,7,8,9,10,11,12,13 Deformities of the head can be divided into abnormalities of the cranium and abnormalities of the face. The term craniofacial deformity is preferred when both the face and the cranium are involved.
The most commonly encountered ophthalmic disorders belong to the craniofacial dysplasias. The craniosynostoses include Apert’s syndrome, Crouzon’s syndrome, Pfeiffer’s syndrome, and other isolated suture abnormalities such as plagiocephaly. The clefting syndromes include cleft lip and palate, oblique facial clefts, and mandibulofacial deformities (Goldenhar’s syndrome, Treacher Collins syndrome, and facial microsomia).
Hypertelorism may present as an isolated anomaly but frequently is a component of syndromes affecting the midface such as frontonasal dysplasia or the craniosynostoses.
CRANIOSYNOSTOSES
Craniosynostosis (craniostenosis) is the premature closure of one or more cranial sutures.14,15,16,17,18,19,20,21 The dysmorphologic makeup observed in craniostenosis is a direct result of premature suture closure and, chronologically, is the initial manifestation. The diagnosis usually is obvious. The malformation varies, depending on the site and the number of sutures involved in the synostosis and on subsequent alterations of growth. In fetuses of 14 weeks’ gestation, that is, about 7.5 cm crown-to-rump length, most of the sutures are formed.22,23 The metopic suture (between the two frontal bones) closes before birth.23 The posterior fontanel normally closes by 2 months of age, the anterolateral suture between 3 and 6 months, and the anterior and posterolateral sutures during the first year of life.23 The sagittal suture closes by 2 years, and the coronal suture closes by 3 years (Fig. 1). The cause of premature suture closure is not known. Either an abnormal fusion of bone centers occurs, or there is malfunction of the underlying dura mater, which normally ensures suture patency.22,23,24,25,26 Subsequent cranial and facial growth are influenced by any prematurely closed suture. Virchow demonstrated in 1851 that, after premature suture closure, growth proceeds parallel to the suture and is inhibited perpendicular to it (Virchow’s law).27 Closure of the sagittal suture results in an elongated skull (scaphocephaly). Single coronal, sagittal, metopic, or lambdoidal suture closure leads to a variety of abnormal shapes such as brachycephaly, scaphocephaly, trigonocephaly, or plagiocephaly (Fig. 2). Classification of craniosynostosis has been divided into three main groups: (1) craniofacial dysostosis (Crouzon’s syndrome), (2) acrocephalosyndactyly (Apert’s syndrome), and (3) simple or single craniosynostosis (plagiocephaly). Classification systems can be confusing because of the overlapping clinical and genetic manifestations.3,4,5 Although a functional classification based on pathomorphogenic features can be used, diagnosis by syndrome has been the traditional method of classification.7 The most commonly encountered craniofacial synostoses are Crouzon’s and Apert’s syndromes.
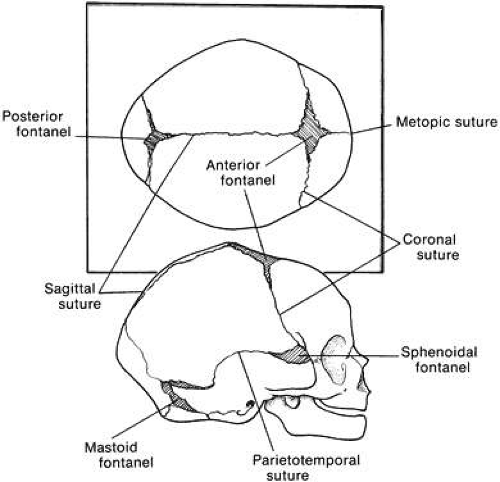 Fig. 1. Position of sutures in a normal full-term infant. |
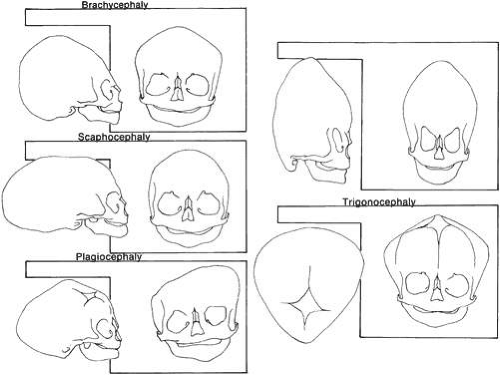 Fig. 2. Skull shapes resulting from premature suture closure. |
Crouzon’s Syndrome
Crouzon’s syndrome originally was described in 1912 in a mother and her daughter under the name of a hereditary craniofacial dysostosis.20 It is characterized by craniostenosis of the acrobrachycephalic or oxycephalic type with associated hypoplasia of the maxilla and exophthalmos. A progressive craniosynostosis often is observed at birth and usually is completed by 2 to 3 years of age. Although there is variable presentation with multiple suture involvement and no characteristic calvarial shape, 96% to 98% of patients show radiographic evidence of coronal and sagittal sutural fusion. This pattern of fusion leads to a more predictable skull shape, which features a shortened calvarium, steep forehead, and flattened occiput6,15,16,17,19,22,28,29 (Fig. 3). Neurologic complications of early suture closure occur commonly and include headaches (29% to 50%), increased intracranial pressure, mental retardation (13%), and seizures (11.5%).17,19 The most characteristic facial feature is midface retrusion, hypoplasia, and shallow orbits.29,30 Brachycephaly, in combination with hypoplasia of the frontal, sphenoid, ethmoid, and maxillary bones, is mostly responsible for these features.
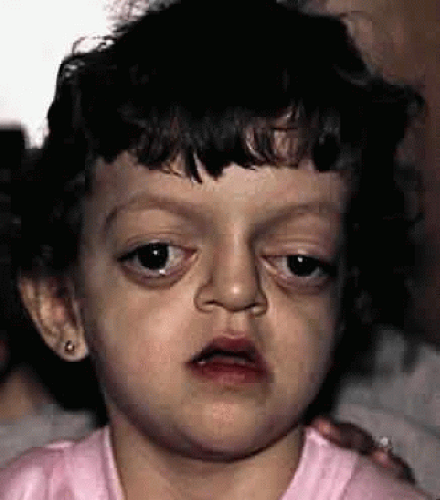 Fig. 3. Patient with typical craniofacial features of Crouzon’s syndrome. |
Hypertelorism and exophthalmos are universal features of all variants of Crouzon’s syndrome. The exophthalmos is secondary to midfacial hypoplasia, resulting in shallow, widespread orbits. One theory states that premature suture closure results in increased intracranial pressure and preferential brain growth in the anterior and middle cranial fossae.24 Another, based on examination of affected fetuses and animal studies, suggests that cranial base changes with resultant stresses initiate osteogenic overstimulation and aberrant bony growth in the calvarium and face.13
Crouzon’s syndrome can affect all ocular structures. Shallow orbits predispose to proptosis and exposure of the cornea and conjunctiva.16,22,31,32 Optic nerve atrophy and edema are vision-threatening complications and have been reported in as many as 80% of patients with this syndrome.33 However, in a series of 25 patients with Crouzon’s syndrome, only 1 had optic nerve disease.32 The most plausible current explanation for optic nerve atrophy is that it is the result of a primary disturbance (hypoplasia or dysplasia) rather than the result of secondary factors (compression atrophy or chronic edema).32,33 Early surgical intervention has resulted in decreased morbidity, although the mechanisms involved in secondary optic nerve problems continue to occur.
Other structural abnormalities of the globe reported in patients with Crouzon’s syndrome include aniridia, anisocoria, corectopia, blue sclera, cataract, ectopia lentis, glaucoma, chorioretinal colobomas, megalocornea, and microcornea.3,13,16,17,22,28,31
Strabismus is present in as many as 92% of patients with Crouzon’s syndrome.32,34,35,36,37,38,39 V-pattern exotropia is the most common misalignment.32,34,39 The V-pattern and hypertropia on adduction usually are secondary to overaction of the inferior oblique muscles, which occurs as a result of an absent or ineffective ipsilateral superior oblique muscle.36,37,38,39 Morax has postulated that the widened, shallow orbits and anomalous muscle insertions change the actions of the muscles.37,38,39 The superior oblique seems to be sagittalized and less effective.33,34,35,36,37,38,39 In a series of 25 patients with Crouzon’s syndrome, 48% had exotropia, 32% had esotropia, and 68% were hypertropic in some position of gaze.32
Patients with Crouzon’s syndrome have large degrees of compound astigmatism, and many also are anisometropic.16,32 Adnexal abnormalities include ptosis, canthal dystopia, nasolacrimal duct obstruction, entropion, and ectropion.31,32,40,41,42,43,44
The advances made in the surgical treatment of Crouzon’s syndrome have changed the pattern and prevalence of visual loss. Structural abnormalities of the globe (e.g., optic atrophy, corneal opacification), which previously were the main causes of vision loss, are less common. The main reason for vision loss is amblyopia. The cause of the amblyopia usually is multifactorial, with contributions from strabismus, uncorrected refractive errors, anisometropia, and ptosis.32
Apert’s Syndrome
In 1896, Apert described this syndrome in a 15-month-old girl.45 He suggested the term acrocephalosyndactyly in a report of nine cases in 1906.46,47 The classification of this syndrome has since been modified and can be confused with other craniofacial syndromes with syndactyly48 (Table 1). This syndrome occurs with a frequency of 1:160,000 births with an autosomal dominant mode of inheritance. Spontaneous mutations have been associated with increased paternal age.46
TABLE 1. Craniosynostosis Syndromes Associated | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
Clinically, the cranium, face, and extremities are involved. Although the cranial features vary, the coronal suture always is affected. The skull is oxycephalic with a flat occiput and steep forehead. The face has a hypoplastic midfacial region with a “parrot-beak” nose. Other features include low-set ears, hypertelorism, and exorbitism.45,46,47 More prominent features of Apert’s syndrome are associated palatal and dental abnormalities, including a high-arched palate, clefting of the soft palate or a bifid uvula, a V-shaped maxillary arch with malocclusion, dental crowding, and supranumerary teeth.47,49 Ear anomalies include pinna defects, conductive hearing loss, and congenital otosclerosis45,46,47 (Fig. 4). Hydrocephalus and mental retardation also occur in these patients.50
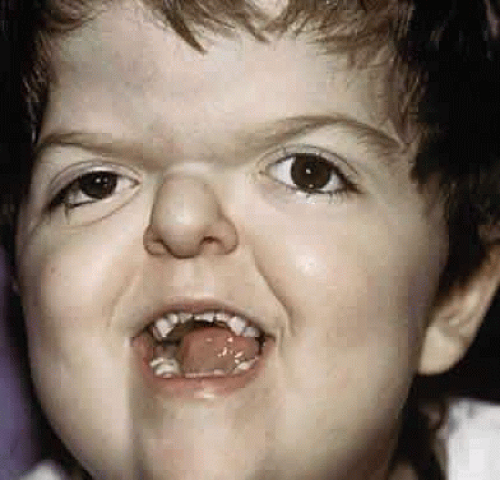 Fig. 4. Patient with typical craniofacial features of Apert’s syndrome. |
Ophthalmic manifestations are similar to those patients described with Crouzon’s syndrome.31,32 Hypertelorism and proptosis are less severe than in Crouzon’s syndrome. Structural abnormalities of the globe also occur in this syndrome and include keratoconus, congenital glaucoma, ectopia lentis, cataracts, exposure keratitis, and optic atrophy.30,31,44,45 Strabismus is common, occurring in 70% to 100% of patients.32,36,39 Exotropia with a V-pattern predominates, although V-pattern esotropia is common. Patients also can have nystagmus, dissociated vertical deviations, and incomitant vertical deviations. The structure of the extraocular muscles is abnormal both microscopically and grossly.38 Anomalous or missing muscles, especially the superior rectus and superior oblique, occur commonly in these patients.31,33,36,37,38,39,51 Refractive errors, in particular, high degrees of oblique astigmatism (from 60% to 100% of patients), are common, as is anisometropia.31,32,34
Adnexal abnormalities include ptosis, nasolacrimal duct obstruction, canthal dystopia, entropion, and ectropion.31,32,41,42,43,44 The high prevalence of visual loss (up to 57% of eyes) in this population of patients results from amblyopia for the same multifactorial reasons as in Crouzon’s syndrome.32
The major associated systemic malformation is syndactyly of the hands and feet. This is most commonly a middigital hand mass involving the second, third, and fourth digits (Fig. 5). Other bony abnormalities include aplasia or ankylosis of the elbow, shoulder, or hip and cervical stenosis.13,46,47
 Fig. 5. Middigital hand mass typical of the syndactyly present in Apert’s syndrome. |
Other Craniostenosis Syndromes
A variety of other craniostenoses (Table 2) share common features with Crouzon’s and Apert’s syndromes but occur less frequently. Pfeiffer’s syndrome is autosomal dominant and has craniosynostosis with oxycephaly, maxillary hypoplasia, hypertelorism, proptosis, depressed nasal bridge, beaked nose, highly arched palate, crowded teeth, hydrocephalus, seizures, and, occasionally, mental retardation.13,52 Ophthalmic features are similar to Apert’s and Crouzon’s syndromes. Distinguishing features include broad, long thumbs and great toes.
TABLE 2. Clinical Characteristics of Craniofacial Synostoses | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Saethre-Chotzen syndrome is characterized by a more variable cranial vault with coronal synostosis, plagiocephaly, scaphocephaly, and oxycephaly. Other features include low-set frontal hairline, facial asymmetry, variable brachydactyly and syndactyly, low-set ears, hearing loss, highly arched palate, and dental abnormalities. Intelligence usually is normal, but mental retardation has been reported.11,13,22
Ophthalmic manifestations of Saethre-Chotzen syndrome differ slightly from Apert’s or Crouzon’s syndrome because of less severe maxillary and midfacial hypoplasia. Hypertelorism and exorbitism are less common. Ptosis is much more common in this syndrome, as well as strabismus and nasolacrimal dysfunction. Amblyopia and optic atrophy again contribute to visual loss in these patients.32
Patients with plagiocephaly exhibit a cranial asymmetry caused by a premature closure of the coronal sutures in one half of the skull. The deformities may be subtle. The skull is flattened in the frontal region on the affected side, often with a compensatory expansion on the opposite side. There is retrusion of the orbital arch on the affected side with a raised eyebrow. The maxillary bone and ear may be affected, with abnormal dental apposition and a downward and forward displaced ear.
Up to 68% of patients with plagiocephaly have ophthalmic involvement. This includes strabismus, cranial nerve palsies, optic neuropathy, ptosis, canthal dystopia, and nasolacrimal duct dysfunction.52,53 Although esotropia is common in these patients, an unusually high prevalence of clinical underaction of the superior oblique muscle ipsilateral to the stenotic suture is present. This manifests with a head tilt and hypertropia on the affected side, which increases on contralateral gaze and ipsilateral head tilt. Often there is underaction of the ipsilateral superior oblique and overaction of the ipsilateral inferior oblique muscle.54 This is postulated to result from a “sagittalization” of the superior oblique because of the orbital deformity.39,44,54 This condition is treated in the same manner as a superior oblique palsy. If the superior oblique muscle is found to be redundant or “loose,” a “tuck” of the tendon is necessary.
Carpenter’s syndrome, first described in 1901, is an autosomal recessive craniosynostosis involving many sutures with preaxial polysyndactyly of the feet and variable brachydactyly and syndactyly of the hands. Mental retardation is uniformly present. Congenital heart disease, short stature, obesity, and lower limb abnormalities are other systemic associations. Hypertelorism, epicanthal folds, and telecanthus are reported ocular abnormalities.19,55,56,57 Other less common craniostenosis syndromes are described in Table 2.
CLEFTING SYNDROMES
The most familiar form of clefting syndromes is isolated cleft lip and palate syndromes. These clefts occur early in development before the 17-mm crown-to-rump length (17 to 20 weeks’ of gestation).58 Interruptions in fusion of the developing facial processes during embryonic development possibly are responsible for these clefting syndromes.58,59 There are five facial processes: a frontonasal, two maxillary, and two mandibular. There may be insufficient outgrowth of the facial processes, poor cellular adhesion resulting in an abnormal epithelial plate, or abnormal cell degeneration of the epithelial plate. The defects of bone, musculature, and soft tissues result from poor fusion of the facial swellings. Failure of mesodermal penetrators with subsequent epithelial dehiscence in the developing facial process as it reaches the midline also is a postulated mechanism of soft tissue defects.7,9,13,58,59
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


