Ocular Pharmacology of Antiviral Agents
Deborah Pavan-Langston
Although it is difficult to target viral components without hitting a few cellular components on the way, important advances have been made in antiviral chemotherapy. Nine systemic or intracameral antiviral drugs (vidarabine, acyclovir, famciclovir, valaciclovir, ganciclovir, valganciclovir, cidofovir, foscarnet, and fomivirsen) and four topical agents (idoxuridine, vidarabine, trifluridine, and acyclovir) are in widespread use in ophthalmic viral infections. Topical and oral BVDU and interferon show clinical potential but remain largely experimental.1,2,3,4,5,6,7,8 The organisms targeted most effectively by these drugs are herpes simplex virus (HSV), herpes varicella-zoster virus (VZV), cytomegalovirus (CMV), vaccinia, and to some extent, adenovirus and Epstein-Barr virus (EBV).
VIRAL REPLICATION CYCLE
The ideal antiviral agent interrupts the viral replicative cycle at a critical phase without involving the host’s cellular metabolism.8,9 Replicative events follow the general order of adsorption, penetration, uncoating, viral genome transcription leading to production of nonstructural and structural viral proteins, synthesis of components, and release.
Attachment and penetration require interaction between virus-specific structures with cellular plasma membranes. Uncoating is usually done by cellular enzymes but may be affected by virus-specific proteins. Viral DNA moves to the nucleus where it is transcribed into messenger RNA (mRNA) by cellular RNA polymerase, a process that also may be affected by viral-specific proteins. The mRNAs then migrate back to the cytoplasm where they sequentially regulate synthesis of virus specific proteins. Key nonstructural proteins (enzymes) include virus-specific thymidine kinase, DNA polymerase, DNase, and ribonucleotide reductase. Synthesis of the DNA viruses of ophthalmic interest occurs in the nucleus, catalyzed by virus-specific DNA polymerase. Capsids and nuclear material are assembled and, in some cases (herpesviruses), host-derived nuclear membranes are acquired before migration of mature organisms to the cell surface for release.
Numerous sites target viral specific functions throughout the replicative cycle. Current antiviral therapy and research focuses on these functions, targeting components as varied as enzymes more specific to virus than cell to virus–specific gene fragments (Table 1).
Table 1. FDA-Approved Antiviral Agents for Ocular Disease | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
IDOXURIDINE
STRUCTURE
Idoxuridine (IDU), the pyrimidine antimetabolite 5-iodo-2′-deoxyuridine, which was introduced commercially in 1962, was the first drug used to control human viral disease.10,11 Structurally, IDU is a thymidine analog with the 5′-methyl group replaced by iodine (Fig. 1 A and B).
MECHANISMS OF ACTION
IDU is incorporated as a thymidine analog preferentially into viral DNA over cellular DNA, as the former replicates more rapidly in infected cells8,10,11 (see Table 1). The drug is not selectively activated but is phosphorylated by both viral and cellular kinases. The IDU triphosphate also inhibits the virus-specific DNA polymerase more than it inhibits the host cell polymerases. IDU may interact specifically with dTMP synthetase or it may be incorporated directly into DNA as IDU monoposphate.12 Substitution of IDU for thymidine in the DNA chain leads to abnormal transcription and translation, thus producing defective viral progeny. Its incorporation into some host DNA adds to the toxicity of the agent.
Although IDU and its phosphorylated metabolites inhibit a variety of cellular enzymes, IDU action as an antiherpetic agent may also occur at a viral biosynthetic level prior to incorporation into the viral genome.10 Phosphorylation of IDU to the triphosphate form (IDUTP) allows this compound to mimic deoxythymidine triphosphate (dTTP), and subsequently IDUTP may exert either an allosteric or a feedback inhibitory effect at the level of dT kinase, deoxycytidine monophosphate deaminase, or cytidine diphosphate (CDP) reductase, ultimately terminating viral replication.12
TOXICITY
The toxic effects of IDU are magnified in tissues undergoing rapid cellular DNA synthesis.13 Clinical trials using the drug systemically for HSV encephalitis revealed that the drug was minimally effective and highly toxic.14 IDU is teratogenic, mutagenic, and potentially carcinogenic. For these reasons it is not used as a systemic agent.
In the eye, adverse reactions to topical IDU include contact dermatitis, punctate epithelial keratopathy, follicular conjunctivitis, lacrimal punctal stenosis and occlusion, and lid margin keratinization.15 IDU is the most toxic of the commonly used topical antiviral drugs, interfering primarily with stromal healing and causing toxic epithelial changes.16
PENETRATION AND PHARMACOKINETICS
In rabbits treated with 0.5% IDU ointment, the drug did not penetrate the intact cornea.17 In human ocular penetration studies, IDU, as the parent nucleoside, did not enter the aqueous humor. Instead, uracil, a metabolic breakdown product of IDU, was detected in the aqueous. Only in patients in whom the corneal epithelium was damaged did IDU penetrate the cornea. When administered systemically (intravenously, intramuscularly, or subcutaneously), the drug is dehalogenated and rapidly metabolized; 50% to 75% is excreted within 4 to 5 hours following systemic administration in animals.18,19
EFFICACY
IDU is used topically and only for HSV keratitis. In vitro studies demonstrated that IDU did not affect the adsorption of HSV in tissue culture cell monolayers, nor did it alter the infectivity of extracellular herpesvirus. IDU did stop viral replication after it had begun and resulted in at least a 2-log unit diminution in viral titers 24 hours post treatment.20,21 The antiviral efficacy of IDU was substantiated in numerous controlled clinical trials.22,23 Healing rates vary from about 75% in complicated cases to 90% in straightforward dendritic or geographic keratitis. When 0.1% IDU was instilled into the eyes of patients with dendritic lesions, relief was noted within 12 to 24 hours after initiation of therapy. By 72 hours, marked healing of the epithelium occurred. IDU is highly effective in treating infectious corneal epithelial herpetic disease but is ineffective in herpetic-induced iritis, stromal keratitis, and other forms of HSV intraocular infection.22,24 It is also less effective than trifluridine and vidarabine if topical corticosteroids are being used concomitantly.25,26
IDU use has diminished with the advent of newer, more convenient drugs and is no longer commercially marketed. Recommended therapy when used is 0.1% q1–2h daily and 0.5% IDU ointment HS for 14 days.
RESISTANCE
HSV resistance to IDU is through modification of the thymidine kinase (TK) gene. Clinically resistant virus strains isolated from patients or created by serial passage through IDU-containing medium all had the common finding of reduced TK activity, sometimes as low as 5.6% of that of the wild-type or parental strain.27,28 IDU-resistant HSV showed cross-resistance to BVDU and acyclovir, intermediate resistance to trifluridine, and full sensitivity to vidarabine and ganciclovir.26
TRIFLURIDINE
STRUCTURE
Trifluridine (TFT; F3T; trifluorothymidine) (5-trifluoromethyl-2′-deoxyuridine, 2′-deoxy-5-[trifluoromethyl] uridine) is a fluorinated pyrimidine nucleoside. Structurally, it is an analog of the deoxyribonucleoside thymidine and is virtually identical to IDU with the exception of three fluorine atoms attached to a methyl radical replacing the iodine.29 (see Fig. 1C). As a pyrimidine nucleoside analog, trifluridine inhibits DNA viruses, and because of its structure it is incorporated into rapidly growing host cell types (e.g., bone marrow).
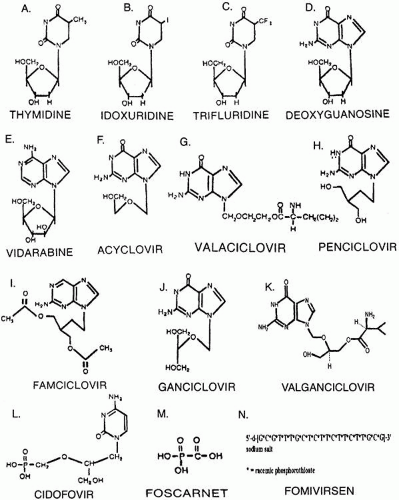 Fig. 1. Structural formulas of major ocular antiviral agents. |
MECHANISM OF ACTION
Although the specific mechanism of trifluridine inhibition of active herpesvirus production is not entirely certain, its antiviral activity appears similar to that of IDU and results from the effect of the drug on viral DNA synthesis29 (see Table 1).
Trifluridine can exert its inhibition at several stages in the viral biosynthetic pathway leading from 2-deoxyuridine-5′-monophosphate to DNA synthesis. Trifluridine acts as a competitive inhibitor of thymidine.30 The drug is initially phosphorylated to trifluridine monophosphate (TFT-MP), which is a potent inhibitor of thymidylate synthetase.31 Following phosphorylation to the triphosphate form (TFTTP), the compound competitively inhibits incorporation of thymidine triphosphate into viral DNA.
TOXICITY
Trifluridine is cytotoxic, teratogenic, mutagenic, and potentially carcinogenic and is sufficiently toxic that it is not used systemically.32,33
In vivo treatment of normal rabbit corneas with trifluridine caused no adverse effects or evidence of corneal toxicity.34 However, when rabbit corneas with standardized epithelial defects were treated with either 1% of 0.1% trifluridine drops eight times a day for 8 days, pathologic changes in the regenerating epithelium were observed. Stromal wound healing appears to be affected by trifluridine therapy (tensile strength was significantly reduced 12 days after wounding and treatment with 1% trifluridine).35
PENETRATION STUDIES AND PHARMACOKINETICS
The biphasic solubility profile of trifluridine enhances the transport of the intact active drug across the cornea. The mechanism of penetration (as for IDU and vidarabine) appears to be by nonfacilitated diffusion demonstrated by linear penetration kinetics through excised, perfused rabbit corneas and the lack of demonstrable saturation kinetics.36 Trifluridine penetrates the corneal epithelium more rapidly than IDU and vidarabine. The presence or absence of an intact epithelial cell layer did not significantly alter trifluridine’s distribution. However, the concentration of trifluridine in the aqueous was increased in debrided or damaged corneas, and the rate of penetration was doubled.37 Trifluridine (1%, four times a day) administered to both infected rabbit eyes and the eyes of patients with a history of recurrent herpetic keratitis penetrated the corneal stroma and achieved therapeutic levels (5–18 μg/mL).36,38
The parent compound of trifluridine is hydrolyzed to 5-carboxy-2′-deoxyuridine within 3 to 5 hours at 37°C and pH 7.2.37,39,40 This metabolite has little or no antiviral activity. In vivo, trifluridine is hydrolyzed to 5-carboxyuracil or to 5-carboxyuridine with the loss of the inorganic fluoride. The rate of elimination correlates with the rate of trifluridine metabolism. Monophosphate, diphosphate, and triphosphate metabolites of trifluridine are found in body tissues. The short serum half-life and toxicity of trifluridine (12–30 min) prevent the use of this drug in systemic herpetic infections.
EFFICACY
Trifluridine not only interferes with the replication of HSV-1 and HSV-2 but also has an effect on vaccinia and certain adenoviruses.41 Trifluridine (0.2–1.7 g/mL) inhibits the cytopathic effects of HSV-1 by 50% in plaque reduction assay.42 Plaque formation was reduced by over 98% when HSV-1 grown into Vero cells was treated with 17 μg/mL trifluridine.43 Trifluridine activity in vitro is comparable to that of IDU, and trifluridine is considerably more active on a weight-for-weight basis than is vidarabine. As observed for both IDU and vidarabine, the strain of HSV-1 appears to be of major importance in determining the relative antiviral efficacy. Trifluridine was shown to inhibit five strains of HSV-1 within a narrow range; however, the susceptibility of five HSV-2 strains was variable, with two strains being insensitive at the maximum nontoxic concentration.44
Trifluridine concentrations of 0.01 to 10 μg/mL administered as topical eye drops to experimentally infected rabbit corneas reduced the severity of the corneal herpetic ulcers when compared with saline-treated controls.22 Trifluridine was more potent on a weight-for-weight basis than IDU in the treatment of McKrae strain–induced HSV-1 herpetic keratitis, and when trifluridine and IDU were compared with respect to their ability to eradicate virus from the preocular tear film, no virus was recovered on days 2 and 4 of the 7-day treatment with trifluridine. However, HSV-1 was present in IDU-treated eyes throughout the treatment regimen.22,45 Two days following discontinuation of therapy, rebound virus shedding had occurred in both groups, with virus titers higher than those observed in control, placebo-treated animals. These results indicate that a critical time period exists in an acute herpetic infection, during which time continued presence of the antiviral agent is necessary to control rebound virus shedding, even though infectious virus cannot be detected in the tear film.
Clinical studies comparing topical 1% trifluridine to 0.1% IDU, 3% vidarabine, or 3% acyclovir have shown that, overall, the latter two drugs and trifluridine have efficacy rates between 90% and 95% regardless of whether steroids are in use.7,26,46,47,48,49 IDU efficacy was about 76%, dragged down by apparently less efficacy because of corticosteroid use in some patients or perhaps by its having been in clinical use so long that certain organisms had become resistant or patients had become allergic to the agent. Although trifluridine had a slight edge over all other drugs in the face of concomitant corticosteroid therapy, no statistical difference could be shown.
VIDARABINE
STRUCTURE
Vidarabine (9-β-D-arabinofuranosyladenine, ara-A) is a purine arabanosyl nucleoside structurally similar to deoxyguanosine51 (see Fig. 1 D and E).
MECHANISM OF ACTION
Vidarabine is not virucidal nor does it prevent attachment or penetration of infectious virus particles into host cells. Like all current antivirals it is virostatic (see Table 1). Its biologic activity is due in part to intracellular phosphorylation, first to the monophosphate (Ara-A-MP) form and subsequently to diphosphate and triphosphate (Ara-A-DP, Ara-A-TP) forms by cellular enzymes. Vidarabine inhibits viral DNA synthesis by several mechanisms. Direct incorporation into DNA as Ara-A-MP results in DNA chain termination. The triphosphorylated form, Ara A-TP, inhibits terminal deoxynucleotidyl transferase and DNA polymerase (α and β). Ara-A-DP and Ara-A-TP also inhibit the activity of ribonucleotide reductase enzymes.52,53,54
TOXICITY
Following systemic or topical administration, vidarabine is converted to a hypoxanthine arabinoside derivative (ara-HX); this metabolite has only 20% of the antiviral potency of the parent compound.55 The toxicity of vidarabine, administered systemically or topically, is negligible over a wide range of pharmacologic dosages. A major problem in systemic administration is that the compound’s relative insolubility requires the use of large volumes of fluid, which can tax cardiovascular and renal homeostatic mechanisms. The drug is also teratogenic and mutagenic and has carcinogenic potential.56 Thus, vidarabine is used systemically only as an alternative to acyclovir or ganciclovir in HSV or VZV drug-resistant, life-threatening situations.
Vidarabine administered topically to rabbits did not impair wound healing as measured by planimetry and histopathologic examination. However, stromal wound strength of vidarabine- or IDU-topically treated corneas was less than that of wounds treated with placebo ointment.16
PENETRATION AND PHARMACOKINETICS
Ocular penetration studies have shown that topically applied vidarabine crosses the cornea in minor amounts in corneas with intact epithelium. The deaminated semiactive metabolite Ara-HX was found in the aqueous in significant amounts following topical vidarabine treatment.17,37 Application of 3% vidarabine in a water-miscible cream produced corneal levels of 20 μg/mL, whereas the same concentration in petrolatum ointment produced levels of only 4.5 μg/mL. Subconjunctival injection in dosages of either 25 or 100 mg resulted in intraocular Ara-HX titers of 6.5 μg/mL and 8.0 μg/mL, respectively.57,58 No significant difference in aqueous Ara-HX concentration was observed among eyes receiving 25 or 100 mg vidarabine.
Following intravenous administration, vidarabine is rapidly deaminated by adenosine deaminase to the less effective Ara-HX form or other noneffective metabolites. Plasma levels of ara-HX are directly related to the rate of infusion. Plasma half-life is approximately 3.5 hours, and the metabolite Ara-HX is well distributed in tissues. Erythrocyte levels of Ara-HX are equal to plasma levels. Ara-HX readily crosses the blood-brain barrier and attains concentrations within 35% of plasma levels in the cerebrospinal fluid.55,59 To enhance the antiviral efficacy of vidarabine therapy, potent inhibitors of adenosine deaminase that stop the conversion of Ara-A to Ara-HX have been used. These inhibitors have enhanced both the cytopathic and the cytotoxic activity of vidarabine in vitro.60,61 Systemic administration of these compounds reduced vidarabine deamination (i.e., increased parent nucleoside concentration), increased cellular adenosine triphosphate (ATP) concentrations, and increased the virucidal activity by 20-fold, but also enhanced the cytotoxicity of these drugs.
EFFICACY
Vidarabine has a broad antiviral spectrum that includes many DNA viruses (HSV-1, HSV-2, VZV, CMV, vaccinia, and pseudorabies virus); it has little or no effect against RNA viruses.62 Plaque reduction assays using several different herpesvirus strains indicated a high degree of strain variability. As is the case with IDU, the cell type, virus passage, and virus strain all significantly affect the antiviral activity. Vidarabine is highly effective against IDU-resistant HSV-1 and HSV-2 in vitro.63
In the eye, topical vidarabine 3% ointment has been found effective in herpetic keratitis and keratouveitis in both animal models and in humans.25,46,47,64,65 In one study of 69 patients with external ocular herpetic keratitis, vidarabine proved to be equal to IDU in reducing tear-film viral titers and in promoting corneal reepithelialization.66 Vidarabine was significantly less toxic than IDU and caused healing of herpetic lesions clinically resistant to IDU. Other studies in patients with extensive dendritogeographic ulcerations substantiate these findings.66 Fewer treatment failures occurred with vidarabine (9.5%) compared with IDU (18.8%) in a study of more than 300 ocular herpes patients.25 Several studies referenced in this section and in the section on trifluridine indicate that there is no statistically significant difference between topical vidarabine, trifluridine, and acyclovir therapy for HSV46,47,49,67
Systemic vidarabine has been effective in therapy for varicella and for herpes zoster ophthalmicus, although it is now in a far fourth place behind valaciclovir, famciclovir, and acyclovir in this regard. Vidarabine therapy (10 mg/kg/day for 5 days) for varicella in immunocompromised patients reduced fever duration, lesion count, and systemic morbidity significantly compared to placebo68 and resulted in more rapid cessation of viral shedding and increased healing rate of lesions.68 Post-herpetic neuralgia could not be evaluated. Its role in TK-negative, acyclovir-resistant HSV mutants continues to make this a drug of interest.3,70,71,72
RESISTANCE
Resistance to vidarabine appears related to viral DNA polymerase rather than to TK. Mutant, vidarabine-resistant viruses with changes in the DNA polymerase gene have been isolated.73,74 This resistance was mapped to an 0.8 kb region within the pol gene and makes viral DNA polymerase less susceptible to Ara-A-TP.73,75 Although other gene products may be affected, most HSV mutations have been found to code for the carboxyl-terminal portion of the viral DNA polymerase.76 Such DNA polymerase mutation is of clinical significance because mutant HSV is resistant not only to vidarabine but also to the other common agents that use this antiviral mechanism. Resistance to vidarabine is uncommon and is discussed with reference to its use in place of HSV and VZV resistant to other antivirals (e.g., IDU, acyclovir).
ACYCLOVIR
STRUCTURE
Acyclovir (ACV; acycloguanosine) (9-[2-hydroxyethoxymethyl]guanine) is a synthetic compound that was designed to mimic substrates for model enzyme systems. Acyclovir is an analog of guanosine or deoxyguanosine in which the 2′- and 3′-carbon atoms are missing (see Fig. 1F). Acyclovir was the first compound engineered to have selective in vitro and in vivo antiviral activity.42,77
MECHANISM OF ACTION
The major modes of action of acyclovir are viral DNA chain termination and rapid inactivation of viral DNA polymerase. Acyclovir is phosphorylated (activated) to acyclovir monophosphate (ACV-MP) specifically by herpesvirus-encoded thymidine kinase, thereby largely bypassing activation in any but infected cells42,77 (see Table 1). This markedly reduces toxicity and increases specificity. ACV-MP is further phosphorylated to acyclovir diphosphate and triphosphate by viral and cellular enzymes. Acyclovir triphosphate (ACVTP) competes for deoxyguanosine triphosphate (dGTP), the natural substrate for virus-specific DNA polymerase and is incorporated irreversibly as ACVMP onto the growing viral DNA chains, thus causing chain termination.78,79 Termination occurs because the acyclovir lacks the 3′ hydroxyl group needed to react with incoming nucleotides. DNA polymerase then binds irreversibly to the acyclovir-terminated chain, and the entire enzyme complex is metabolically inactive. Little to no inactivation of cellular polymerase occurs.42,78,79,80 Elion and co-workers have determined that ACVTP inhibits HSV DNA polymerase (DNA nucleotidyl transferase) ten to 30 times more efficiently than cellular DNA polymerase, another factor in reducing toxicity.77 Approximately a 3000-fold concentration increase (above that which is virucidal) is needed to inhibit host cell growth.
TOXICITY
Since acyclovir is activated specifically by herpesvirus-coded TK, it was anticipated that the drug would be relatively nontoxic. Following corneal application of 3% acyclovir ointment, no toxic corneal effects were noted, and the quality and rate of re-epithelialization and stromal wound strength of the cornea were not impaired.16 Clinical study did note rare diffuse punctate keratitis that cleared following discontinuation of acyclovir therapy, but this apparent toxicity was thought to be a function of the drug vehicle.65
The only important metabolite of acyclovir is 9-carboxymethoxy-methyl guanine (CMMG), an inactive compound that accounts for up to 14% of acyclovir dosage administered to humans.81 Renal clearance of acyclovir ranges from 75% to 80% of the total body clearance and is substantially greater than the clearance of creatinine, indicating that glomerular filtration and tubular secretion mechanisms (possibly the organic acid secretory system) are involved.82 The drug may rarely cause renal failure when given in high dose (>5 mg/kg) intravenously if there is renal insufficiency or dehydration. Hydration of all patients should be monitored and patients with renal insufficiency should have reduced doses and prolongation of the intervals between dosing to minimize this risk. Rare central nervous system toxicity, delerium tremens, and coma have been reported.83 Other common side effects are gastrointestinal distress and headache. There is no evidence that acyclovir is a carcinogen or teratogen, and at therapeutic doses, there is no effect on the hematopoietic or immune systems.84
PENETRATION AND PHARMACOKINETICS
Acyclovir can be administered safely and effectively by intravenous or subconjunctival injection, orally, or topically. Its adsorption following oral administration is variable and appears to be species dependent.81,84,85,86 Animal studies have indicated that following oral administration in mice, 50% of the acyclovir is absorbed, and peak plasma concentrations occur 2 to 4 hours following ingestion.82
In humans, oral absorption of acyclovir is incomplete, with bioavailability of the drug ranging between 15% and 30% of the dosage.87 Peak plasma concentrations occur 1.5 to 2.5 hours following administration, and steady-state plasma concentrations following multiple oral doses of 2.5, 5, 10, and 15 mg/kg administered every 8 hours were 6.7 μg/mL, 9.7 μg/mL, 20 μg/mL and 20.6 μg/mL, respectively. These values are similar to the peak plasma concentrations observed following equivalent single oral doses, and acyclovir does not accumulate in plasma following repetitive dosing. Oral dosing of 200 mg q4h reaches steady-state levels ranging from 1.4 to 4.0 μM, which is inhibitory for HSV-1 and HSV-2; however, doses of 800 mg 5 times per day are needed to yield peak and trough serum levels, respectively, of 6.9 μM and 3.5 μM to inhibit most strains of VZV.88,89,90
Studies on the concentrations of acyclovir in the tear film and aqueous in patients on receiving doses of 400 mg 5 times daily showed levels of 3.28 μM (0.96–8.79 μM) and 3.26 μM (1.10–5.39 μM), respectively, four hours after the last oral dose.91,92 The mean ED50 (effective dose reducing viral plaque count in tissue culture by 50%) of HSV-1 ranges from 0.15 to 0.18 μM and 0.1 to 1.6 μM, repectively, for the tear and aqueous levels, which indicated that the levels achieved were well in excess of that which should be needed to eliminate the virus.8,88,91,92,93 This is not always the case. Stromal HSV particles may persist in the face of prolonged, therapeutic doses of oral acyclovir.94 Drug resistance may have played a role in this finding. In comparison to HSV, the inhibitory doses for VZV are much higher at 3 to 4 μM, resulting in the need for fourfold higher drug dosing, as noted above, and less leeway in terms of resistance.93
Following topical instillation of 3% acyclovir ointment in the inferior cul-de-sac of 25 eyes (every 5 hours for four to six doses prior to cataract extraction), the mean acyclovir concentration in the aqueous humor was 1.7 μg/mL, which falls within the therapeutic range.95 Cutaneous adsorption of acyclovir may occur through damaged skin, but systemic adsorption is limited.
Total body distribution of intravenous14 C-acyclovir has been studied in several animal species.86 Acyclovir rapidly enters all tissues following administration in both mice and rats. In humans with normal kidney function the serum half-life is about 3 hours. Intravenous dosing of 5 mg/kg three times daily results in serum ID50 levels (inhibitory dose reducing viral plaque count in tissue culture by 50%), well above that needed for HSV-1 and HSV-2 at all times; however, dosage of 10 mg/kg three times daily was necessary to avoid trough levels that fell below the ID50 of several VZV strains.86,88,96,97 Multidose intravenous therapy with 400 mg to 1200 mg/M2 every 8 hours resulted in acyclovir concentrations in kidney, lung, and brain that were 1000%, 131%, and 25% to 70% of plasma levels, respectively.98 Cerebrospinal fluid (CSF) acyclovir levels were approximately 50% of corresponding plasma levels. The rapid penetration of acyclovir into the CSF has made this drug an important compound for treating focal and disseminated central nervous system herpetic infections.
Intravitreal acyclovir levels in humans 2 hours after administering13 mg/kg intravenously were at inhibitory levels for HSV-1 and HSV-2, VZV, and EBV.147 Regular dosing of 5 mg/kg three times daily yielded concentrations of 8.8 to 11.0 μM, well in excess of the inhibitory dose for HSV-1 and HSV-2, VZV, and EBV, as did 25 mg of subconjunctival acyclovir.99
EFFICACY
In vitro studies have demonstrated that acyclovir has a broad antiviral spectrum, including HSV-1 and HSV-2, VZV, EBV, and, to a lesser extent, CMV. Acyclovir is 160 times more potent than vidarabine and ten times more potent than IDU.100,101 The inhibitory doses for various viruses have been noted previously.
Acyclovir (100 μg/mL) has been used in a drug-induced suppression model of HSV-1 infection in trigeminal ganglion cells. In this system, it produces a suppressed HSV-1 infection that is functionally identical to HSV-1 latency in vivo. Acyclovir has also been shown to suppress the reactivation of latent virus in explanted trigeminal ganglia.102 Latency was not eradicated, however, as demonstrated by an increase in viral titer following acyclovir removal. The rabbit eye model of HSV-1 infection has been used extensively to assess the in vivo efficacy of acyclovir.103,104
primary genital HSV (oral or IV)
recurrent genital HSV in immunocompetent patients (oral)
mucocutaneous HSV in immunocompromised patients (oral or IV)
HSV encephalitis (IV)
neonatal HSV (IV)
varicella in immunocompetent (oral) or immunocompromised patients (IV then oral)
Herpes zoster in immunocompetent (oral) or immunocompromised patients (IV then oral)
Possibly EBV infections (oral)
Multiple clinical studies on infectious HSV epithelial keratitis comparing topical 3% acyclovir with 3% vidarabine ointment, 0.1% IDU drops, or 1.0% trifluridine drops in recommended doses revealed no statistically significant difference among the four drugs, although there was a trend suggesting that acyclovir was superior to IDU if concomitant corticosteroids were given. Acyclovir ophthalmic ointment is not commercially available in the U.S., although it is marketed in other countries.1,49,65,89,90,91,108,109,110,111
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


