Fibroblastic or myofibroblastic tumors make up the majority of soft tissue tumors of the aerodigestive tract and range from nonneoplastic or benign proliferations (fibromas) to high-grade malignancies (undifferentiated high-grade pleomorphic sarcomas). Distinguishing between these tumors is important because they have vastly different prognoses (
Table 11.2). Such distinctions cannot always be made with small biopsy specimens, although high-grade malignancies can usually be distinguished from lower grade neoplasia and reactive conditions.
“Fibroma”
A variety of benign fibrous lesions that may or may not be neoplastic can be found within the upper aerodigestive tract and have been termed
“fibromas.” These include nasal and oral fibromas (traumatic/irritation fibromas, peripheral ossifying fibromas, cementifying fibromas, etc.). One should be cautious regarding the use of the term fibroma within the upper aerodigestive tract outside of these lesions.
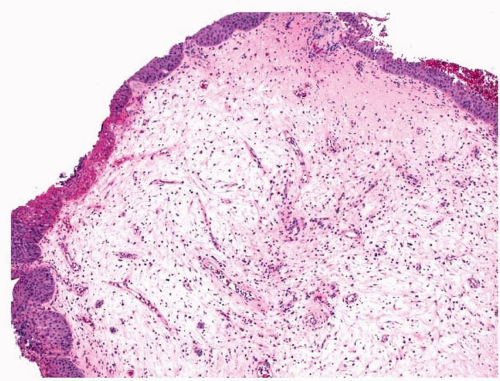
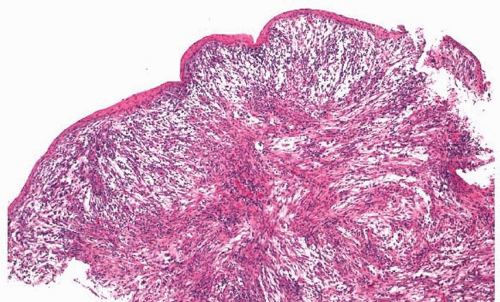
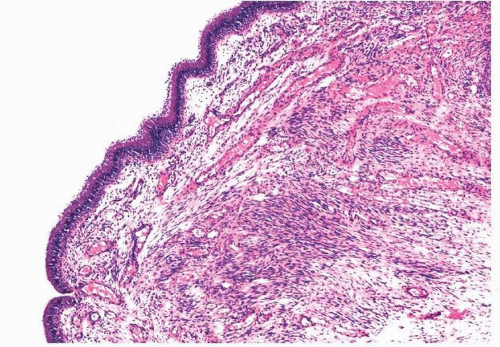
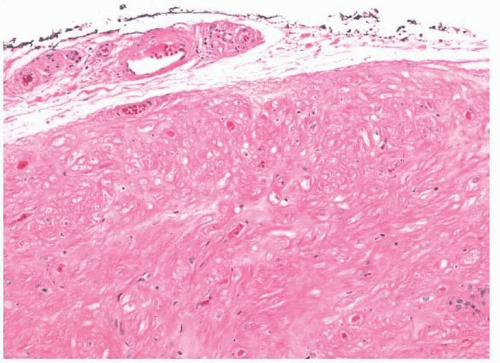
Nasal fibromas or fibrous polyps are small, smooth polypoid lesions that develop in or near the nasal vestibule.
2 They are usually less than 1 cm in size and frequently present incidentally. Histologically, mature fibroblasts are arranged haphazardly within dense collagen (
Fig. 11.1, e-
Figs. 11.1 and
11.2). More than mild cytologic atypia is not seen and mitotic figures are usually not present. The lesions are benign and do not recur after resection.
A number of different lesions occur in the mouth, which have been termed
fibroma.3,4 These lesions all appear to be reactive, yet have a few differences between them.
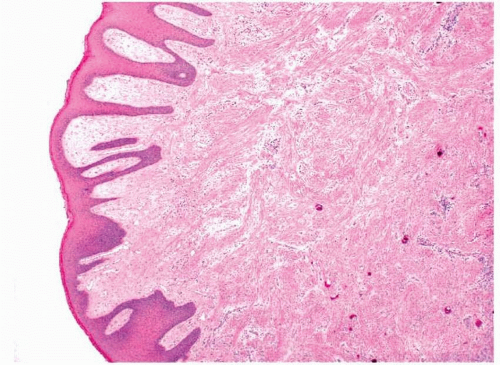
Traumatic or
irritation fibromas are the most common lesions. These are polypoid lesions that are composed of subepithelial, haphazardly arranged, bland fibroblasts within a collagenous stroma. The overlying squamous epithelium can be normal or can show changes consistent with continued trauma such as parakeratosis or acanthosis. The lesions can be ulcerated and chronically inflamed
(epulis fissuratum), may have large, stellate fibroblasts
(giant cell fibroma), and may contain odontogenic epithelium
(retrocuspid papule).4 Some mesenchymal cells of the gingiva may have a pluripotent nature and be able to differentiate into either osteoblasts or cementoblasts; thus, some apparently reactive fibromas within the mouth can form osteoid or cementum
(peripheral ossifying fibroma or
peripheral cementifying fibroma, respectively) (
Figs. 11.2 and
11.3, e-
Fig. 11.3).
4 (Ossifying fibroma of the bone is discussed in
Chapter 12.)
Nodular Fasciitis
Nodular fasciitis and its related lesions usually affect the subcutaneous soft tissue.
5,6,7 and
8 These lesions have been reported rarely in the mouth and very rarely in the nasal cavity and one should be wary of diagnosing them elsewhere in the upper aerodigestive tract.
9,10 They are reactive proliferation and characteristically affect young adults. Clinically, they present as
rapidly enlarging, solitary, and often tender masses and are usually less than 2 cm in size. The lesions do not recur if they are completely resected.
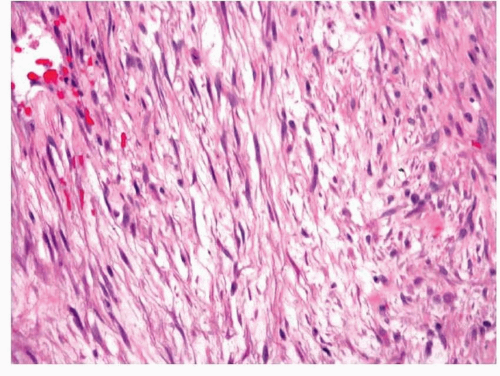
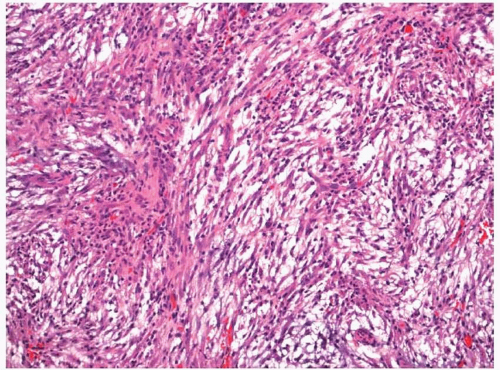
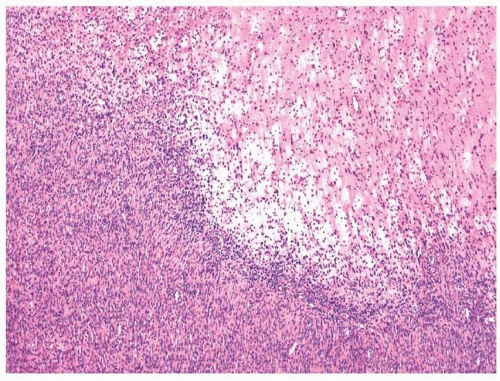
Nodular fasciitis is well circumscribed; however, some minor degree of infiltration into the surrounding skeletal muscle can be present.
5 The tumors are composed of proliferating fibroblasts and myofibroblasts arranged in a haphazard or somewhat storiform appearance in a background loose, somewhat myxoid stroma (
Fig. 11.4, e-
Fig. 11.4).
5,6 and
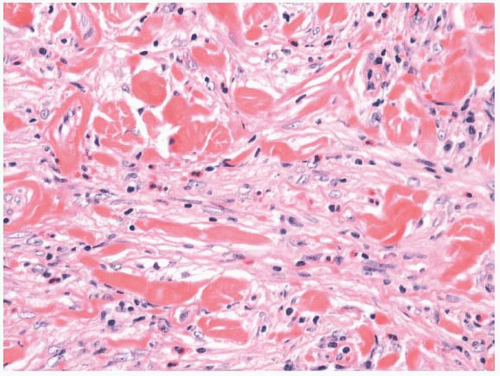
7,10 Zonation, a characteristic feature consisting of alternating degrees of cellularity within single intermediate power fields, is usually present. The fibroblasts may be plump but usually show only minimal cytologic or nuclear atypia (
Fig. 11.5). Older lesions may have a more collagenized stroma (e-
Fig. 11.5). The number of mitotic figures varies greatly from case to case but can be high. Background mixed inflammatory cells and extravasated red cells are usually seen (e-
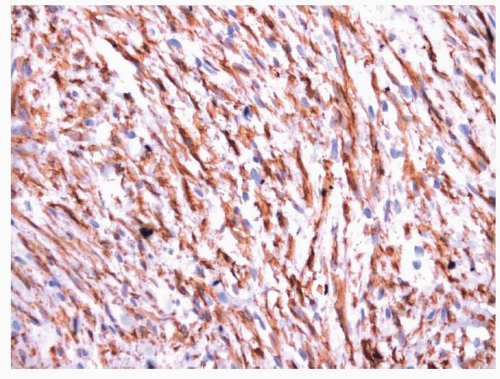
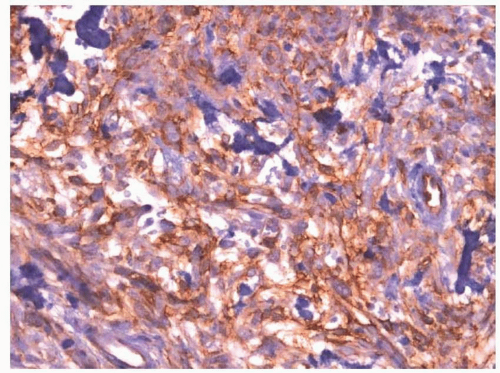
Fig. 11.6). As with other myofibroblastic lesions, the spindled cells are typically immunoreactive with antibodies to smooth muscle actin (SMA) (
Fig. 11.6).
As nodular fasciitis is neither aggressive nor malignant, it is important to distinguish it from other fibroblastic or myofibroblastic lesions of the area such as fibromatosis and inflammatory myofibroblastic tumor (
Table 11.2). Fibromatosis usually does not present as a rapidly growing mass and is not well circumscribed. Furthermore, while some areas of fibromatosis may be more proliferative, the tumors are generally much more collagenized. Immunostaining with antibodies to β-catenin may also be helpful (see below). Distinguishing nodular fasciitis from inflammatory myofibroblastic tumors on small biopsy specimen may be impossible. Inflammatory myofibroblastic tumors can appear more inflamed and
have more sclerotic areas; however, they often have areas very reminiscent of nodular fasciitis. Clinical history and immunostaining with antibodies to anaplastic lymphoma kinase 1 (ALK1) (see below) may be helpful.
Fibromatosis
Aggressive or desmoid-type fibromatosis can involve the head and neck and may occasionally be sampled in biopsies from the upper aerodigestive tract.
2,11,12 In one study, involvement of the neck was noted in
roughly 20% of the extra-abdominal desmoid tumors studied.
11 The tumors can occur at any age and are somewhat evenly distributed throughout the first five decades of life. Extra-abdominal desmoids occur slightly more often in women, although they are not usually associated with pregnancy. The tumors frequently recur, especially when wide excision is not an option (some have noted that sinonasal tract fibromatoses recur less commonly than other extra-abdominal fibromatoses).
12 Although the tumors do not, by definition, metastasize, they can cause death due to the local destruction and entanglement of vital structures. This is especially true in the head and neck. Fibromatoses are neoplastic and cytogenetic abnormalities can frequently be identified, especially involving the long arm of chromosome 5.
13 This is interesting, as abdominal fibromatoses have long been recognized to occur with familial adenomatous polyposis (FAP, Gardner’s syndrome), a hereditary disorder characterized by mutations of the
adenomatous polyposis coli (APC) gene, which is located on the long arm of chromosome 5.
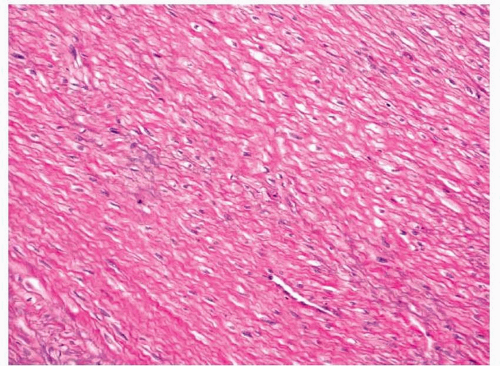
14,15Grossly, the tumors are white-tan and rubbery and appear whorled and infiltrative.
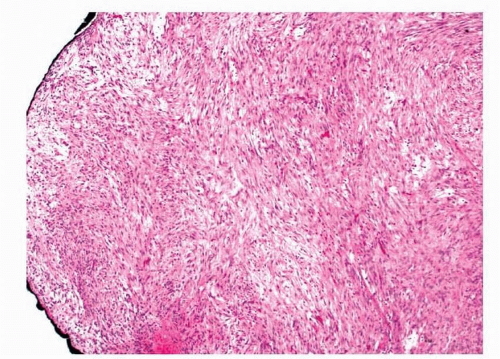
2,11,12 Histologically, they are composed of interlacing bundles and fascicles of spindled to plump myofibroblasts (
Figs. 11.7 and
11.8, e-
Figs. 11.7 and
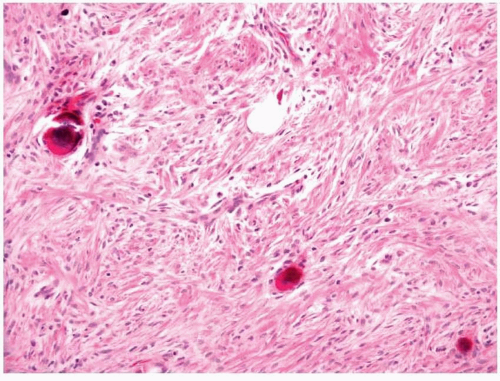
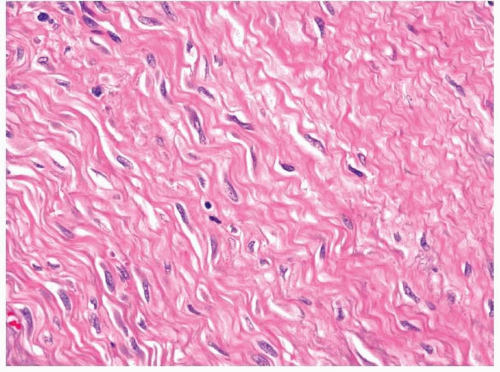
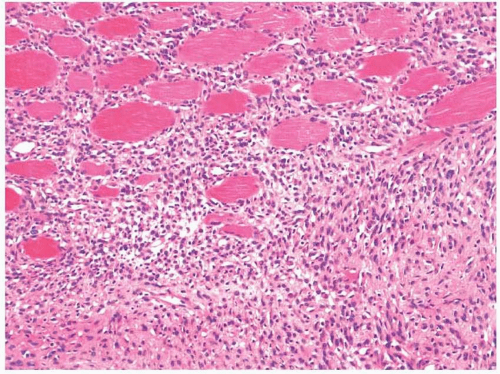
11.8). Collagen deposition is usually seen and can vary in amount within a tumor, thus rendering some areas hypocellular in appearance, whereas other areas appear moderately cellular. The margins of fibromatoses are infiltrative and fascicles of spindled myofibroblasts can be seen dissecting through adjacent soft tissues. Frequently, entrapped skeletal muscle can be seen with degenerating changes, and it is important
not to confuse these changes with epithelial or multinucleated giant cells (
Fig. 11.9, e-
Fig. 11.9). Occasional mitotic figures may be present; however, more than mild cellular or nuclear atypia should exclude the diagnosis. The neoplastic cells of fibromatoses have an immunophenotype similar to other myofibroblastic tumors and are immunoreactive with antibodies to vimentin and SMA. Focal desmin immunoreactivity can be observed. It has recently been noted that most of these tumors, regardless of whether they actually have
β-catenin or
APC mutations, will show nuclear localization of β-catenin by immunohistochemistry, and that this can be helpful for distinguishing these tumors from other fibroblastic or myofibroblastic tumors.
16
Myofibroma/Myofibromatosis
Myofibromas are uncommon tumors that have a marked predilection for the head and neck and are somewhat more common in boys and men.
17,18,19,20 and
21 The tumors can be solitary or multifocal (myofibromatosis). Most solitary myofibromas and cases of myofibromatosis occur in children, although both can be diagnosed at any age. These tumors are most often identified in the mouth when they involve the upper aerodigestive tract. They are generally benign and have even been noted to regress. Multifocal tumors that involve vital structures can sometimes lead to the death of the patient.
Myofibromas can vary in size and are circumscribed but not encapsulated, with a whorled or lobulated cut surface.
17 Microscopically, the tumors are composed of haphazardly arranged, interweaving bundles and
short fascicles of plump spindled cells (
Fig. 11.10).
17,18,19 and
20 The cytoplasm is pale and eosinophilic and can rarely have small vacuoles. The spindled cells have oval, rounded, or tapered nuclei with vesicular chromatin and small, indistinct nucleoli (
Fig. 11.11). The tumors often have hypocellular and hypercellular areas with many small slitlike vessels. In the more cellular areas, the vessels may display a hemangiopericytoid appearance,
often with the intravascular polypoid projection of tumor (e-
Fig. 11.10). Occasional mitotic figures may be noted (up to 5 per 10 hpf). Despite their gross appearance of circumscription, infiltration of the surrounding tissue is usually present, with entrapped soft tissue elements (e.g., peripheral nerves or skeletal muscle) (e-
Fig. 11.11). Multinucleated giant cells can be present, as can degenerative myxoid changes and necrosis (e-
Figs. 11.12 and
11.13). Some cases have abundant stromal collagen.
Immunohistochemically, the tumor cells express SMA and musclespecific actin (MSA) (e-
Fig. 11.14).
17,18,19 and
20 Antibodies to desmin are usually nonreactive. Focal reactivity with antibodies to S100 protein has been described, but most tumors are nonreactive. These tumors need to be distinguished from other myofibroblastic lesions that can be seen with biopsy (
Table 11.2). They lack the inflammatory background of inflammatory myofibroblastic tumors and of nodular fasciitis, and the less cellular areas of myofibromas do not have the loose appearance of either of these tumors. The alternating hypocellular and hypercellular areas seen with these tumors are not present in low-grade myofibroblastic sarcomas or fibromatoses, at least not to the same degree.
Solitary Fibrous Tumor/Hemangiopericytoma
Many sinonasal tract tumors originally described as hemangiopericytomas appear to be more closely related to glomus tumors (see below).
22 In fact, once such tumors are reclassified, true solitary fibrous tumors (or hemangiopericytomas as understood elsewhere in the soft tissues) of the upper aerodigestive tract are found to be much less common than they were once believed to be. Nonetheless, small series of these tumors within the
sinonasal area and the mouth have been published, while a few scattered case reports have described these tumors in the larynx.
23,24,25,26,27 and
28 The tumors arise submucosally in adults of either sex and may appear either as lumps or as polypoid masses. Solitary fibrous tumors are mostly benign, but some (10% to 15% of solitary fibrous tumors and up to 30% of tumors classified as hemangiopericytomas) will behave aggressively and may recur or even metastasize.
29The current WHO classification scheme for soft tissue neoplasms combines solitary fibrous tumors and hemangiopericytomas into a single section and then discusses both individual entities while noting that there is marked clinicopathologic overlap between the two entities.
29 These tumors are characteristically well-circumscribed rubbery lesions, although circumscription would obviously be difficult to assess with the piecemeal specimens removed from the sinonasal tract.
23,24,25 and
26,29 They usually have admixed hypercellular and hypocellular areas and may have focal myxoid change. The neoplastic cells are bland, spindled to ovoid, and haphazardly arranged and have oval to polygonal nuclei, with fine chromatin and inconspicuous nucleoli. These cells are intertwined between collagen fibers of varying thicknesses (
Fig. 11.12, e-
Fig. 11.15). Mitotic activity can be present but is usually low (less than 2 mitotic figures per 10 hpf), and necrosis should not be seen. In fact, when increased mitotic activity (>3 mitotic figures per 10 hpf), necrosis, marked cytologic atypia, or infiltrative margins are seen, the tumors are more likely to behave malignantly.
29,30 and
31 Many thin-walled, small to medium-sized vessels are present that often have a prominent “staghorn” appearance and can focally have
a thickened, collagenous cuff (e-
Fig. 11.16). Mast cells are usually present. While some entrapped normal tissue may be present, the lesions rarely infiltrate bony tissues (e-
Fig. 11.17).
Immunohistochemically, solitary fibrous tumors stain similarly and react strongly with antibodies to vimentin, CD34, and bcl-2 (
Fig. 11.13).
23 Some staining, albeit weak, is often seen with antibodies to CD99. Antibodies to specific vascular and muscle markers are usually nonreactive (weak, focal reactivity with antibodies to SMA may rarely be seen). The differential diagnosis of solitary fibrous tumors is broad, and the diagnosis is often considered one to be made after the exclusion of numerous other entities. In the sinonasal area, glomangiopericytomas must be excluded. This is easily accomplished by immunostaining the tumors with antibodies to CD34 and SMA. At all sites, other tumors, such as synovial sarcomas, fibrosarcomas, myofibroblastic tumors, and various neural tumors, must be excluded (
Table 11.1). Unlike synovial sarcomas, solitary fibrous tumors do not express TLE1.
32
Inflammatory Myofibroblastic Tumor
Extrapulmonary inflammatory myofibroblastic tumors occur throughout the body. A little more than 10% of these involve the upper aerodigestive tract.
33,34,35,36 and
37 The tumors can be found in patients of all ages, but children are more frequently affected. Within the upper aerodigestive tract, they present with the nonspecific symptoms of a mass lesion; however, clinical features can include fever and anemia. Infrequently, patients may be found to have thrombocytosis or hypergammaglobulinemia. Some inflammatory myofibroblastic tumors have rearrangements of
ALK on the short arm of
chromosome 2.
38 The tumors occasionally recur (approximately 20%); however, death from the disease is rare.
Grossly, the tumors can range from fleshy to firm and may have hemorrhage, necrosis, or calcification.
33,34 The histologic features seen with these tumors can be quite variable and have been generally described as having three different patterns, all of which may be seen in any particular tumor. One pattern resembles granulation tissue or nodular fasciitis and is composed of loosely arrayed, stellate to plump spindled cells with abundant eosinophilic cytoplasm (
Figs. 11.14 and
11.15, e-
Fig. 11.18). The cells are embedded within a myxoid or edematous stroma with numerous small blood vessels, a mixed inflammatory infiltrate, and extravasated red cells. Mitotic figures are common but atypical forms are not present. Another pattern is more compact, with the myofibroblasts having a fascicular or storiform growth pattern. Plasma cells are abundant with this pattern and typical mitotic figures can frequently be found (e-
Figs. 11.19 and
11.20). The third pattern is characteristically less cellular with dense collagen (
Fig. 11.16). Fewer mitotic figures are identified in these areas and the inflammatory infiltrate tends to be less prominent. Calcifications can sometimes be seen in these areas.
Immunohistochemically, the myofibroblasts show immunoreactivity with antibodies to vimentin, SMA, and MSA (e-
Fig. 11.21).
33 Limited reactivity with antibodies to desmin can be seen; however, tumor cells have not been found to be immunoreactive with antibodies to myoglobin.
39 Immunoreactivity with antibodies to cytokeratin is seen in up to 36% of the cases. Consistent with the activation of
ALK, approximately 30% can be found to overexpress ALK1 by immunohistochemistry.
40These tumors can be distinguished from other myofibroblastic tumors by their characteristic inflammatory infiltrate and variable growth
patterns. Occasionally, ALK1 immunostaining may be helpful, especially when the tumor expresses the protein; however, some have shown ALK1 staining not to be unique to inflammatory myofibroblastic tumors.
40 The large spindled cells with eosinophilic cytoplasm may occasionally appear reminiscent of the rhabdomyoblasts or strap cells seen with embryonal rhabdomyosarcomas. Cross-striations and less differentiated areas should
not be seen with inflammatory myofibroblastic tumors nor should immunoreactivity with antibodies to myogenin. The characteristic inflammatory cell infiltrate seen with inflammatory myofibroblastic tumors is not usually seen with embryonal rhabdomyosarcomas.
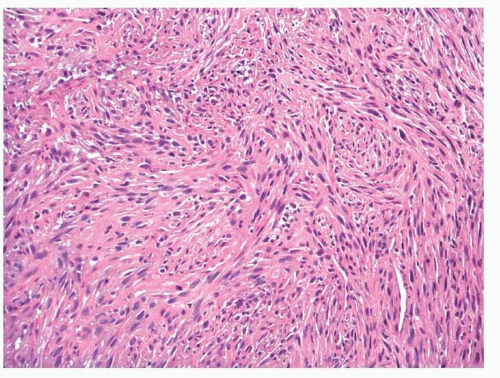
Low-Grade Myofibroblastic Sarcoma
Myofibroblastic differentiation is common in soft tissue neoplasms. The diagnosis of low-grade myofibroblastic sarcoma is used to connote what is believed to be a specific clinicopathologic entity.
41,42 In the two largest reports discussing these lesions, they have been noted to occur more frequently in men and have been limited to adults (although some case reports discuss younger patients). Approximately a quarter of these tumors have been found to develop in the mouth.
43 The tumors recur in approximately 40% of cases; however, less than 10% have been found to metastasize.
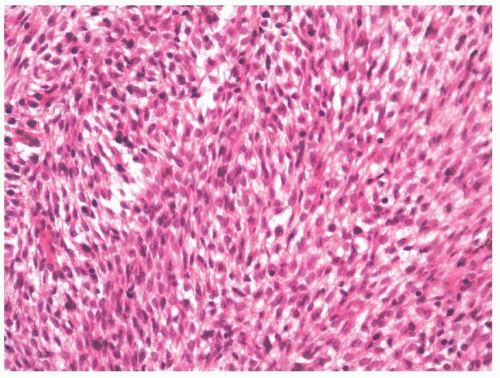
Low-grade myofibroblastic sarcomas are firm and fibrous and most have ill-defined margins; however, they can appear distinctly circumscribed.
41,42 Histologically, the tumors usually appear to infiltrate the surrounding soft tissues. Neoplastic cells are arranged in fascicles of varying lengths; however, herringbone, hemangiopericytoid, and storiform growth patterns have been noted. Cellularity can vary, but areas of hypercellularity are always found (
Figs. 11.17 and
11.18, e-
Figs. 11.22 and
11.23). Tumor cells are spindled and have pale, eosinophilic cytoplasm with tapered nuclei (e-
Fig. 11.24). The nuclei have fine or vesicular chromatin and inconspicuous nucleoli. Most cases have little atypia, although
occasional examples display moderate atypia with enlarged, pleomorphic, hyperchromatic nuclei and prominent nucleoli. Occasional stellate cells with elongated cell processes may be seen. Mitotic activity is present and up to 10 mitotic figures per 10 hpf have been found. Tumor necrosis may be noted focally in rare cases.
Immunohistochemically, these tumors react with antibodies to vimentin and SMA and frequently with antibodies to desmin (e-
Fig. 11.25).
41,42 They do not react with antibodies to cytokeratins or S100 protein. Most do not react with antibodies to CD34, although occasional cases may have limited weak staining. This immunohistochemical staining pattern of these tumors should help to distinguish them from other spindle cell lesions, such as neural tumors, solitary fibrous tumor, sarcomatoid carcinoma, and malignant melanoma. It is obviously not very helpful for distinguishing the tumors from smooth muscle tumors or other myofibroblastic proliferations. Low-grade myofibroblastic sarcomas have more atypia than leiomyomas and they have infiltrating borders. Also, the fascicular cell arrangement of leiomyomas is much more pronounced and the individual cells of these tumors are more eosinophilic with blunted rather than tapered nuclei. Leiomyosarcomas also have more eosinophilic cells and often have a more prominent fascicular appearance. They typically have more cytologic atypia than low-grade myofibroblastic sarcomas.
Distinguishing low-grade myofibroblastic sarcomas from the various other myofibroblastic lesions of the upper aerodigestive tract may be particularly challenging (
Table 11.2). Low-grade myofibroblastic sarcomas should be more cellular and show more cytologic atypia than fibromatoses,
and fibromatoses are much less frequently immunoreactive with antibodies to desmin. Myofibromas have more plump cells and typically show less cytologic atypia than low-grade myofibroblastic sarcomas. Although the cellularity of low-grade myofibroblastic sarcomas can vary, the typical biphasic cellularity seen with myofibromas is not seen with these tumors. Like fibromatoses, myofibromas infrequently show immunoreactivity with antibodies to desmin. Finally, inflammatory myofibroblastic tumors typically have a more pronounced inflammatory cell infiltrate and have areas with looser stroma than low-grade myofibroblastic sarcomas. They also frequently have
ALK rearrangements and express ALK1 by immunohistochemistry.
44
Fibrosarcoma
Fibrosarcomas have been reported to involve the upper aerodigestive tract, especially the sinonasal area.
2,45,46 and
47 Most patients are adults, although rare cases of infantile fibrosarcomas have been reported. Adult-type fibrosarcomas typically arise in middle-aged or older individuals. Patients present with nonspecific complaints such as pain, obstruction, or bleeding. Destruction of adjacent bone is often seen by imaging. The tumors often metastasize, especially to the lungs, and the 5-year survival rate is approximately 50% for adult-type fibrosarcomas that are grade 2 or higher.
47Most adult-type fibrosarcomas are highly cellular and usually show only limited collagen production (although it is typically present, at least focally) (
Fig. 11.19, e-
Fig. 11.26).
2,48 Less cellular areas may, however, be seen within a tumor. The elongated spindled cells are arranged in bundles that often intersect to form the classic “herringbone” pattern (Fig. 11.20,
e-
Fig. 11.27). Most tumors have rather uniform cells with spindled to oval nuclei and little pleomorphism. Mitotic figures are usually abundant. Immunohistochemically, fibrosarcomas will react with antibodies to vimentin and may have limited reactivity with antibodies to SMA.
49Adult-type fibrosarcomas need to be distinguished from a number of lesions including the other fibroblastic and myofibroblastic tumors discussed in this chapter (
Tables 11.1 and
11.2). In general, fibrosarcomas are typically more cellular and mitotically active than benign or the lower grade malignancies. Low-grade fibrosarcomas that are difficult to distinguish from fibromatoses and that may have a better prognosis than higher grade (grade 2 or above) fibrosarcomas (e-
Fig. 11.28) have been reported at this site. Some have suggested using a mitotic count to distinguish these lesions, designating tumors with 6 to 10 mitotic figures per 10 hpf as “borderline” and those with more than 10 as fibrosarcomas. β-Catenin immunostaining may also be helpful. Other spindle cell tumors, especially monophasic synovial sarcomas and malignant peripheral nerve sheath tumors, need to be distinguished from these malignancies. This can usually be accomplished with immunohistochemistry (
Table 11.1). Adult-type fibrosarcomas typically have limited cellular atypia, and more pronounced atypia should lead to a diagnosis of pleomorphic sarcoma. Finally, as with all spindle cell sarcomas of the upper aerodigestive tract, these tumors need to be distinguished from sarcomatoid carcinomas, especially after radiation therapy for squamous cell carcinoma. Intraepithelial neoplasia (squamous dysplasia) or focal squamous differentiation can often be identified with sarcomatoid carcinomas, and many sarcomatoid carcinomas show immunoreactivity with antibodies to cytokeratins.
The diagnosis of adult-type fibrosarcoma is now made only after other, better defined entities have been excluded.
50 With this in mind, previous reports of fibrosarcoma of the upper aerodigestive tract should be interpreted with some caution, especially as there appears to be a significant overlap between what some might call fibrosarcoma and others might call malignant peripheral nerve sheath tumors.
46 Recently, some have described a low-grade sarcoma of the sinonasal tract that can have limited myogenic and S100 immunoreactivity, having both histologic and immunohistochemical features that overlap with these tumors.
51
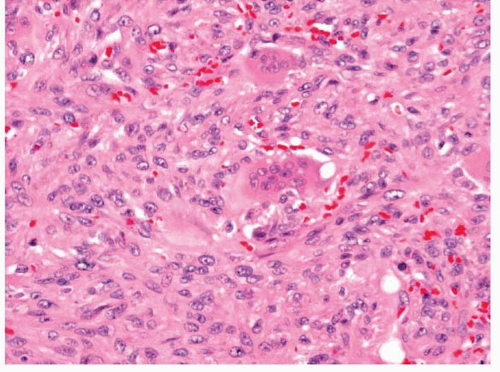
Undifferentiated High-Grade Pleomorphic Sarcoma
Undifferentiated high-grade pleomorphic sarcomas (malignant fibrous histiocytomas) usually involve the extremities, but have been noted within the upper aerodigestive tract.
45,52,53,54 and
55 The tumors occur in older adults, often in the sixth and seventh decades of life. Once other lesions such as sarcomatoid carcinomas or dedifferentiated liposarcomas are distinguished from these tumors, it appears that they have an overall poor prognosis, with a 5-year survival rate of 50% to 60%.
45 Interestingly, some of these lesions involving the head and neck have been reported after radiotherapy, which has also been suggested as a risk factor for the transformation of conventional squamous cell carcinomas to a sarcomatoid carcinoma.
56Histologically, these tumors have a variable appearance but are characterized by marked cytologic and nuclear pleomorphism (
Fig. 11.21, e-
Fig. 11.29).
52,53,54 and
55 Many tumor cells will be spindled, but large round and polygonal tumors cells are often noted with atypical multinucleated tumor
cells. Some cells will have abundant eosinophilic cytoplasm and some may appear finely vacuolated. Cellularity is usually high, although some tumors have a myxoid or fibrous background and appear less cellular. Mitotic figures are abundant and necrosis and hemorrhage are common. Osteoclastlike giant cells are usually present and can be numerous
(undifferentiated high-grade pleomorphic sarcoma with giant cells) (
Fig. 11.22, e-
Figs. 11.30 and
11.31). Background inflammation may be present and can occasionally be prominent
(undifferentiated pleomorphic sarcoma with prominent inflammation).The diagnosis of undifferentiated high-grade pleomorphic sarcoma is one of exclusion. In the upper aerodigestive tract, a sarcomatoid carcinoma must first be ruled out. Immunostaining with antibodies to cytokeratin can be helpful, although a lack of staining does not exclude sarcomatoid carcinoma. A careful search for a squamous component to the tumor or overlying squamous dysplasia should be made and the identification of either should lead one to diagnose the lesion as a sarcomatoid carcinoma. Dedifferentiated liposarcomas and osteosarcomas should be distinguished from these lesions because of their different prognoses, and a careful search for lipoblasts and osteoid should be made. Immunohistochemistry can be used when needed to differentiate leiomyosarcomas and angiosarcomas from these tumors.
Other Myofibroblastic Tumors
Other myofibroblastic tumors have rarely been reported to involve the upper aerodigestive tract, and these include
dermatofibromas or
benign fibrous histiocytomas, dermatofibrosarcomas, giant cell angiofibroma, infantile fibrosarcomas, and
sclerosing epithelioid fibrosarcoma. One should keep in mind that most soft tissue tumors can develop in the head and neck and can, either primarily or through secondary extension, involve the upper aerodigestive tract.