19 Strictly speaking, uveitis is defined as inflammation of the uvea that is composed of the iris, ciliary body, and choroid. Clinically, the term uveitis is used more broadly to define any condition that causes intraocular inflammation. This is because inflammatory processes within the uveal tract frequently spill over into the adjacent anterior chamber, vitreous, and retina. Similarly, inflammatory disorders primarily affecting the retina and sclera may involve the uveal tissues. There are many causes of uveitis, including infections such as syphilis, toxoplasmosis, herpes simplex, and tuberculosis or malignancies like primary central nervous system (CNS) lymphoma and leukemia. Most causes of uveitis are the result of either primary ocular or systemic immune-mediated or autoimmune diseases in which cell-mediated immunity is believed to a play major role. Uveitis ranks as the fourth most common cause of blindness in developing countries, behind macular degeneration, diabetic retinopathy, and glaucoma. The prevalence of uveitis from all causes is estimated at 40 per 100,000, with an annual incidence of about 15 per 100,000.1 Uveitis affecting the posterior segment is responsible for 10% of all cases of blindness in developed countries.2 Although uveitis affects persons of all age groups, it is diagnosed more frequently in patients aged 20 to 40 years. In this age group, uveitis has been reported to equal diabetic retinopathy as the leading cause of blindness in developed countries. Children constitute 5 to 10% of patients with uveitis.3 Many different approaches have been developed in an attempt to organize the various causes of uveitis and to help the practitioner to reach the correct diagnosis when faced with a patient with intraocular inflammation (Table 19–1). Although none of the classification schemes alone is perfectly complete, each has individual merit. When used collectively, they require that the examiner carefully consider the patient’s demographics, history, and clinical findings to generate a differential diagnosis that will direct the diagnostic testing needed to determine the most probable cause of the uveitis. The simplest classification scheme divides the causes of uveitis into infectious and noninfectious. From a practical point of view, this distinction is the most important because it will dictate the approach to treatment with either specific antibiotics or immunosuppressive therapy. A second classification scheme attempts to categorize the various causes of uveitis as acute or chronic on the basis of their clinical course. In another scheme, uveitis is classified as granulomatous if findings consistent with granulomas are present on ophthalmic examination and nongranulomatous when no evidence of ocular granulomas is found. The most widely accepted and used classification scheme for uveitis is the anatomic classification, which divides uveitis into four categories (anterior, intermediate, posterior, and panuveitis), depending on the primary site of inflammation in the eye. Acute uveitis has an abrupt, symptomatic onset of the ocular inflammation in one or both eyes. Although recurrent, each new episode of ocular inflammation or flare spontaneously resolves, and the eye becomes quiet within 6 weeks even in the absence of treatment. Most causes of acute uveitis are either idiopathic or associated with the class I human leukocyte antigen, HLA-B27. Examples of such diseases include HLAB27-associated anterior uveitis, ankylosing spondylitis, and Reiter syndrome. Behçet syndrome and many of the white-dot syndromes are examples of acute posterior segment uveitis.
Noninfectious Inflammation
Categorizing Uveitis
How Is Uveitis Defined?
What Are the Causes of Uveitis?
How Common Is Uveitis?
How Is Uveitis Classified?
How Do You Distinguish between Acute and Chronic Uveitis?
Infectious (diseases caused by parasites, viruses, fungi, bacteria, or mycobacteria) or |
NonInfectious (diseases of immune-mediated or unknown etiology) |
Acute (duration of ocular inflammation less than 6 wk) or |
Chronic (duration of ocular inflammation greater than 6 wk) |
Granulomatous (granulomatous lesions associated with ocular inflammation) or |
Nongranulomatous (ocular inflammation not associated with granulomatous lesions) |
| Anatomic |
Anterior (ocular inflammation primarily affecting the iris, ciliary body pars plicata, anterior chamber, and corneal endothelium) |
Intermediate (ocular inflammation primarily affecting the vitreous, ciliary body pars plana, and peripheral retina) |
Posterior (ocular inflammation primarily affecting the retina and choroid) |
Panuveitis (ocular inflammation involving the entire uvea and adjacent layers without a predominant focus) |
Uveitis is considered chronic if the intraocular inflammation persists for 6 weeks or longer with or without treatment. Most noninfectious causes of uveitis are chronic. Sarcoidosis is the most common cause of chronic posterior segment uveitis. Other types of chronic uveitis that affect the posterior segment are birdshot retinochoroidopathy, serpiginous choroidopathy, multifocal choroiditis, pars planitis, and primary intraocular lymphoma.
How Do You Differentiate between Granulomatous and Nongranulomatous Uveitis?
Uveitic diseases can be classified as nongranulomatous or granulomatous by the absence or presence of specific clinical findings on ocular examination suggestive of the presence of granulomas in the eye. Granulomas are discrete collections of macrophages, epithelioid cells, and giant cells surrounded by chronic inflammatory cells and fibroblasts. In the eye, granulomatous inflammation manifests by the presence either of greasy-appearing or “mutton-fat” keratic precipitates, nodular lesions in the iris stroma (Busacca nodules) or pupillary margin (Koeppe nodules), and lesions subjacent to the retinal pigment epithelium (RPE), known as Dalen-Fuch nodules. Only a limited number of uveitic diseases, such as sarcoidosis, sympathetic ophthalmia, multiple sclerosis (MS), lens-induced uveitis, syphilis, tuberculosis, and the Vogt–Koyanagi–Harada (VKH) syndrome, are known to cause granulomatous inflammation in the eye. Some diseases, such as sarcoidosis, MS, and toxoplasmosis, can present with either a granulomatous or nongranulomatous appearance.
How Do You Define Uveitis Anatomically?
The International Uveitis Study Group has proposed an anatomic classification for uveitis divided into four categories: anterior, intermediate, posterior, and panuveitis.4 According to this classification scheme, the type of uveitis is defined by the location of the primary site of inflammation in the eye. Anterior uveitis is diagnosed when the intraocular inflammation is predominantly limited to the anterior segment or structures, including and anterior to the lens–iris diaphragm. In severe cases of anterior uveitis, inflammatory cells from the anterior chamber may spill over into the retrolental space, producing an anterior vitreitis. Other terms used for anterior uveitis are iritis, iridocyclitis, and anterior cyclitis.5 Some causes of anterior uveitis include the HLA-B27-associated diseases, juvenile rheumatoid arthritis, sarcoidosis, Behçet disease, Fuch heterochromic iridocyclitis, and masquerade syndromes (Table 19–2).
Intermediate uveitis is characterized by inflammation that primarily affects the pars plana, vitreous, and vessels of the peripheral retina. Inflammatory cells originating in the middle segment of the eye typically diffuse throughout the vitreous cavity and may escape into the anterior chamber, producing a concomitant anterior uveitis. Diseases commonly associated with intermediate uveitis are sarcoidosis, inflammatory bowel disease (IBD), MS, and pars planitis (Table 19–3). Other names in the literature that describe patients with intermediate uveitis include vitreitis, peripheral exudative retinitis, cyclochorioretinitis, chronic posterior cyclitis, and peripheral uveoretinitis.6
| Idiopathic |
| HLA-B27 associated anterior uveitis |
| Ankylosing spondylitis |
| Reiter syndrome |
| Psoriatic arthritis |
| Inflammatory bowel disease |
| Juvenile rheumatoid arthritis |
| Fuch heterochromic iridocyclitis |
| Sarcoidosis |
| Syphilis |
| Masquerade syndromes |
HLA, human leukocyte antigen.
| Noninfectious |
| Idiopathic |
| Intermediate uveitis of the pars planitis subtype (pars planitis) |
| Sarcoidosis |
| Inflammatory bowel disease |
| Multiple sclerosis |
| Primary intraocular lymphoma |
| Masquerade syndrome |
| Infectious |
| Lyme disease |
| Syphilis |
| Tuberculosis |
The term posterior uveitis defines ocular inflammatory conditions that primarily affect the retina and choroid. Although the vitreous is not the primary focus of the inflammation in posterior uveitis, vitreitis is typically present as a result of the spillover of inflammatory cells from the retina or choroid into the vitreous cavity. Similarly, in cases of moderate or severe posterior uveitis, inflammatory cells can reach the anterior chamber. Posterior uveitis can be further classified as retinitis if the retina alone is affected, choroiditis if the choroid alone is affected or as retinochoroiditis or chorioretinitis to denote inflammatory conditions affecting both layers simultaneously. Posterior uveitis also can be described as focal or multifocal. Toxoplasmosis and toxocariasis are causes of posterior uveitis typically characterized by a focal lesion, whereas fungal and viral pathogens, sarcoidosis, sympathetic ophthalmic, the VKH syndrome, serpiginous choroidopathy and birdshot retinochoroidopathy characteristically cause multifocal lesions. Retinal vasculitis refers to those causes of posterior uveitis in which the primary focus of inflammatory disease seems to be the retinal vasculature. Many causes of retinal vasculitis are associated with systemic diseases such as Behçet syndrome, systemic lupus erythematosus (SLE), Wegner granulomatosis, and sarcoidosis (Table 19–4). Finally, the term panuveitis is used to describe diseases characterized by intraocular inflammation simultaneously affecting all intraocular structures without a predominant focus in any segment. The most frequently diagnosed causes of panuveitis are syphilis, sarcoidosis, the VKH syndrome, infectious endophthalmitis, Behçet syndrome, and sympathetic ophthalmia (Table 19–5). In this chapter, the term posterior segment refers to ocular structures posterior to the lens–iris diaphragm and the term posterior segment uveitis will include conditions classified anatomically as intermediate, posterior, and panuveitis.
| Noninfectious causes of multifocal posterior uveitis |
Sarcoidosis |
Masquerade syndromes |
Vogt–Koyanagi–Harada syndrome |
Serpiginous choroidopathy |
Birdshot chorioretinopathy |
Sympathetic ophthalmia |
Multifocal choroiditis |
White-dot syndromes |
Multiple evanescent white dot syndrome (MEWDS) |
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) |
Acute retinal pigment epitheliitis |
Acute zonal occult outer retinopathy (AZOOR) |
Punctate inner choroidopathy (PIC) |
Primary intraocular lymphoma |
Masquerade syndromes |
| Noninfectious systemic diseases that cause retinal vasculitis |
Behçet disease |
Sarcoidosis |
Multiple sclerosis |
Crohn disease |
Wegener granulomatosis |
Systemic lupus erythematosus |
Polyarteritis nodosa |
Polymyositis/dermatomyositis |
Postvaccination syndrome |
Buerger disease |
Takayasu disease |
Cogan syndrome |
Sjögren syndrome |
Allergic granulomatosis |
| Noninfectious primary ocular diseases that cause retinal vasculitis |
Idiopathic |
Eales disease |
Intermediate uveitis of the pars planitis subtype |
Birdshot retinochoroidopathy |
Masquerade syndromes |
Signs and Symptoms of Uveitis
Symptoms of Uveitis
The most common symptoms of uveitis affecting the posterior segment are blurred vision, floaters, and visual loss. In some cases, the patient may be asymptomatic. The blurred vision may be the result of protein and inflammatory cells in the anterior chamber or vitreous, cataract, or the sequelae of intraocular inflammation, such as macular edema or hypotony. Floaters can be attributed to aggregates of vitreal inflammatory cells, vitreous syneresis, and detachment of the posterior vitreous, which can produce mobile opacities that cast shadows upon the retina. The major cause of visual loss in patients with posterior segment uveitis is cystoid macular edema. Visual loss also may result from secondary glaucoma, retinal neovascularization with vitreous hemorrhage, retinal detachment, epiretinal membranes, subretinal neovascularization, subretinal fibrosis, and atrophy of the optic nerve, RPE, or choroid. Ocular pain in patients with uveitis can be attributed to inflammation in the iris and ciliary body. Patients with posterior uveitis, however, are often asymptomatic or complain only of mild ocular discomfort. Severe pain is unusual and may be the result of increased intraocular pressure resulting from angle closure, pupillary block, or the development of scleral inflammation (sclerouveitis). Other symptoms, such as photopsias, metamorphopsia, micropsia, macropsia, scotomas, dyschromatopsia, and nyctalopia, may be reported in patients with posterior segment uveitis and in many cases correlate with the presence of lesions in the fundus.
| Syphilis infection |
| Endophthalmitis |
| Sarcoidosis |
| Vogt–Koyanagi–Harada syndrome |
| Behçet disease |
| Sympathetic ophthalmia |
What Are the Signs of Uveitis in the Anterior Segment?
Signs of uveitis in the anterior segment of the eye include ocular injection, band keratopathy, keratic precipitates, aqueous cell and flare, hypopyon, anterior and posterior synechiae, and cataract. Conjunctival injection associated with uveitis can be differentiated from conjunctivitis by the lack of involvement of the fornix and palpebral conjunctiva. Conjunctival injection is a typical sign of acute anterior uveitis but is less common in posterior segment uveitis. Ciliary flush or circumlimbal injection is considered to be an external marker of ciliary body inflammation and a more specific marker of uveitis. Band keratopathy, the subepithelial deposition of calcium salts in the middle third of the cornea, is frequently found in children with chronic uveitis but is infrequently seen in adults. Keratic precipitates, aggregates of inflammatory cells that usually deposit on the lower half of the corneal endothelium, are the most common corneal finding in uveitis. Nongranulomatous keratic precipitates are composed of neutrophils and lymphocytes and appear clinically as punctate, fleck-like, linear, or stellate endothelial opacities. Granulomatous keratic precipitates, composed of macrophages and giant cells, are larger, rounder, and often appear pigmented and greasy, hence the term mutton fat. The presence of keratic precipitates does not define the activity of the intraocular inflammation because they can occur later in the active disease course and may persist after the uveitis has resolved. Anterior chamber cells or inflammatory cells circulating within the aqueous humor are required for the diagnosis of anterior uveitis and panuveitis. Although usually found in most cases of intermediate uveitis, anterior chamber cells may not be found in milder cases of posterior uveitis. Flare, the ability to visualize a beam of light in the anterior chamber, is a manifestation of increased vascular permeability in the anterior segment. Although typically accompanying cells in the anterior chamber, flare alone is not a sign of active inflammation because damaged anterior segment blood vessels may continue to leak proteins into the anterior chamber long after the active inflammation has resolved. A hypopyon, the acute accumulation of inflammatory cells that layer in the inferior angle, is an uncommon finding in uveitis that always should raise the differential of an infectious endophthalmitis, Behçet disease, and HLA-B27-associated uveitis. Synechiae are adhesions between the iris and the anterior chamber angle or cornea (peripheral anterior synechiae) or the pupillary iris and lens (posterior synechiae). Although they can develop within days after the onset of severe intraocular inflammation, synechiae are more typical of chronic or recurrent types of uveitis. Posterior subcapsular cataracts are the most common lens opacity associated with uveitis. Uveitis can be associated with elevated intraocular pressure or hypotony. Increased intraocular pressure can result from trabeculitis, clogging of the trabecular meshwork by inflammatory debris, angle closure, or pupillary blockage caused by peripheral anterior and posterior synechiae, respectively. Hypotony may result from ciliary body detachment caused by a cyclitic membrane or dysfunction due to inflammation.
What Are the Posterior Segment Signs of Uveitis?
The salient clinical signs of the uveitis in the posterior segment are vitreitis, retinal vasculitis, cystoid macular edema, swelling of the optic disc, and the presence of focal or multifocal retinal or choroidal infiltrates. Vitreitis characterized clinically as vitreous cells and haze (a reduction in vitreous clarity) is caused by the infiltration of cells and protein derived from inflammatory foci in the ciliary body, retina, or choroid into the vitreous cavity. The cellular exudate in the vitreous may be localized to the retrolental space (anterior vitreitis) or concentrated above a chorioretinal lesion, but it is more likely to be diffuse within the cortical vitreous. Aggregates of inflammatory cells appearing as grayish white opacities in the vitreous, called snowballs, provide an obvious explanation for the patient’s complaints of floaters. Other common vitreal findings in posterior segment uveitis are vitreous syneresis, posterior vitreous detachments, and vitreous veils (aggregates of inflammatory material adherent to synergetic vitreous strands). On resolution of the uveitis, the vitreous haze rapidly clears, but the cells, snowballs, and veils tend to persist, with many of the remaining cells taking on a pigmented appearance.
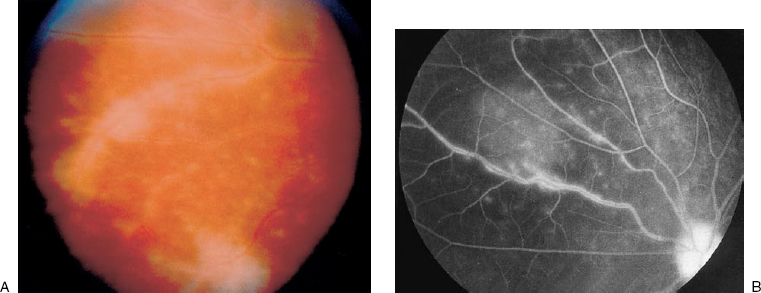
FIGURE 19–1. Retinal vasculitis. (A). Prominent sheathing, the accumulation of inflammatory exudates around retinal vessels, can be seen in this case of severe retinal vasculitis. (B). The fluorescein angiogram shows staining and leakage along the retinal venules characteristic of retinal vasculitis.
Retinal vasculitis is clinically denoted by perivascular sheathing (exudation of inflammatory cells along vascular walls) on ophthalmoscopy and by vascular leakage and staining of the vessel walls on fluorescein angiography (Figs. 19–1 and 19–2). Retinal vasculitis limited to the peripheral or anterior retinal vessels with sparing of the vessels in the posterior pole is characteristic of an intermediate uveitis and may result in perivascular hemorrhages and peripheral retinal ischemia as a result of capillary dropout leading to peripheral retinal neovascularization. Retinal vasculitis in the posterior pole, in contrast, is more likely to induce venous dilatation, splinter hemorrhages, cotton-wool spots, and vascular occlusion that can result in macular ischemia or cystoid macular edema. Generally, retinal vasculitis in the posterior pole can be distinguished from noninflammatory causes of retinal vascular occlusion, like central or branch retinal vascular occlusion by the absence of vascular sheathing and other evidence of intraocular inflammation. Inactive vasculitis is characterized by perivascular sheathing without angiographic leakage and attenuated retinal vessels that may appear sclerotic. Cystoid macular edema, retinal edema, and edema of the optic nerve, intraretinal infiltrates, and exudative retinal detachments all are manifestations of vascular permeability in patients with uveitis.
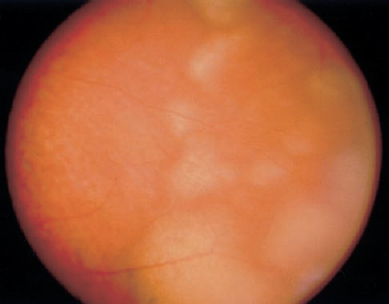
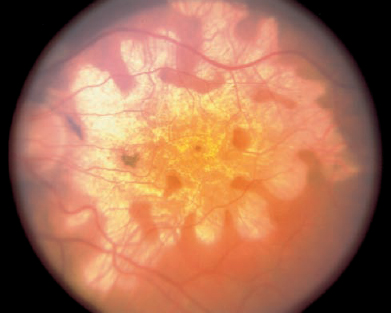
FIGURE 19–2. Primary intraocular lymphoma. Note the creamy appearance and indistinct margins of subretinal tumor masses in the peripheral retina. The lack of clarity of the photograph is due to the grade 1+ vitreous haze.
The presence of retinochoroidal or chorioretinal lesions distinguishes intermediate uveitis from cases of posterior and panuveitis. The chorioretinal or retinochoroidal lesions that define posterior and panuveitis may have a wide variety of appearances. In some cases of posterior uveitis, the recognition of a particular appearance or pattern of distribution by the lesions or their association with certain demographic, systemic, or other ocular findings may allow a specific diagnosis to be made, but there is considerable overlap. Important clinical features to note about the fundus lesions in patients with posterior uveitis are their size, distribution, location, and degree of activity. In general, active lesions have ill-defined margins, appear as creamy with a yellowish coloration, may be associated with retinal edema or opacification, and have no associated RPE hyperpigmentation (Fig. 19–2). In contrast, inactive lesions more often have distinct edges; appear more white or whitish gray because of fibrosis; might be associated with atrophy of the adjacent retina, RPE, and choroidal tissues; and are frequently pigmented as a result of RPE proliferation (Fig. 19–3).
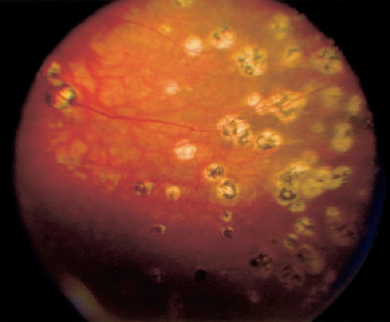
FIGURE 19–3. Multifocal choroiditis. Depicted here are multiple, inactive, variably pigmented “punched-out” chorioretinal lesions that can be seen in a variety of other conditions causing posterior uveitis, including sarcoidosis, the Vogt-Koyanagi-Harada (VKH) syndrome, and sympathetic ophthalmia.
Posterior Segment Uveitis
How Is the Severity of Posterior Segment Uveitis Determined?
The severity of uveitis may be determined not only by the findings on the clinical examination but also by an assessment of the way the ocular disease has responded to previous therapy and the affect of the ocular disease on the patient’s quality of life and daily activities. An objective quantification of the intraocular inflammation during the ophthalmic examination is the most important tool used to determine the severity of the uveitis. In the anterior chamber, cells and flare are recorded, whereas in the vitreous cavity, vitreous cells and haze are recorded. Anterior chamber cells and flare are best seen by directing a 1 mm 1 mm slit lamp beam into the anterior chamber using the highest illumination obliquely across the eye and focusing posterior to the cornea. According to the grading scheme utilized at the National Eye Institute (NEI) for anterior chamber cells a grade of 0 to 4+ is given by counting the number of cells within the light beam7 (Table 19–6). It is important to differentiate white blood cells that signify active inflammation from pigmented cells, such as red blood cells and iris pigment epithelial cells, which also can be seen in the anterior chamber but do not indicate an active uveitis. Flare is proportional to the concentration of protein in the aqueous humor. Clinically, flare is graded on a scale from 0 to 4+ (Table 19–6), the latter representing anterior segment protein leakage so extensive that a fibrin clot is present.7 Fluorophotometry and the laser flare meter are ophthalmic instruments that also can be used to objectively assess the degree of aqueous flare. The quantification of vitreous cells and haze can be more difficult in patients with active anterior uveitis, cataract, or posterior synechiae. Furthermore, the vitreous cells may be localized to only a part of the vitreous cavity. Using the NEI approach (Table 19–7), vitreous cells are graded from 0 to 4+ using a Hruby, 90- or 78-diopter lens at the slit-lamp biomicroscope to view the vitreous cells in retroillumination.7 In the vitreous, haze is believed to be a better indicator of active inflammation than vitreous cells because the degree of haze combines the optical effect of the cellular infiltrate and protein leakage in the vitreous cavity. Our grading scale from 0–4+ for vitreous haze (Table 19–7) includes a trace grade and is based on the view of the optic disc and posterior retina using an indirect ophthalmoscope and 20 diopter lens.8 In assessing the degree of vitreous cells using this method, it is important to correct mentally for corneal and lens opacities and anterior segment inflammation. The use of photographic standards makes this system more reproducible than other subjective grading schemes.8
| Number of cells | Cell Grade |
|---|---|
| 0 | 0 |
| 1–5 | Trace |
| 6–15 | 1+ |
| 16–25 | 2+ |
| 26–50 | 3+ |
| >50 or hypopyon | 4+ |
| Slit beam | Flare grade |
Slit beam is optically undetectable | 0 |
Slight (any appreciable light in slit beam) | 1+ |
Mild (iris and lens details clear through slit beam) | 2+ |
Moderate (iris and lens hazy through slit beam) | 3+ |
Severe (plasmoid aqueous or fibrin present in anterior chamber) | 4+ |
Modified from Nussenblatt RB, Whitcup SM, Palestine AG. Examination of the patient with uveitis. In: Nussenblatt RB, Whitcup SM, Palestine AG, eds. Uveitis: Fundamentals and Clinical Practice. St. Louis: Mosby; 1996:58–68, with permission.
| No. of Cellsa | Description | Grade |
|---|---|---|
| 0–1 | Clear | 0 |
| 2–20 | Few opacities | Trace |
| 21–50 | Scattered opacities | 1+ |
| 51–100 | Moderate opacities | 2+ |
| 101–250 | Many opacities | 3+ |
| >250 | Dense opacities | 4+ |
aCells are counted using a Hruby, 90- or 78-diopter lens.
Modified from Nussenblatt RB, Whitcup SM, Palestine AG. Examination of the Patient with Uveitis. In: Nussenblatt RB, Whitcup SM, Palestine AG, eds. Uveitis: Fundamentals and Clinical Practice. St Louis: Mosby; 1996:58–68, with permission.
The use of ancillary ophthalmic tests also can be helpful in determining the severity of uveitis in the posterior segment and may aid in the discrimination of the various uveitic diseases. Fluorescein angiography is valuable for the detection of retinal and choroidal infiltrates, vascular staining and leakage, retinal, optic disc and cystoid macular edema, retinal nonperfusion and neovascularization, and subretinal neovascular membranes. Indocyanine green (ICG) angiography is useful for revealing the extent of choroidal inflammatory lesions. B-scan ultrasonography and ultrasound biomicroscopy (UBM) can demonstrate choroidal thickening, retinal and vitreous detachments, vitreous inflammatory debris, pars plana exudates, particularly when the posterior pole cannot be viewed because of posterior synechiae and media opacities. Additionally, ultrasonography is beneficial for the interpretation of intraocular masses. Electrophysiologic studies, such as the visual evoked potential (VEP), can help to differentiate optic nerve disease from retinal damage resulting from chronic intraocular inflammation; electroretinography (ERG) and electrooculography (EOG) generally show nonspecific reduction in cases of uveitis corresponding to the extent of chorioretinal disease.
The patient’s subjective view of his or her ocular condition should be elicited when the history is taken. Previous medications given to treat the uveitis, the affect of the medications on the intraocular inflammation, and the course of the disease once treatment is tapered or withdrawn should be carefully recorded. It is also important to know how the patient tolerated the recommended therapy, specifically whether corticosteroids were prescribed. This information, together with an accurate assessment of the objective clinical findings, determines the severity of the uveitis in a given patient and forms the basis for recommending an appropriate tailored therapeutic regimen.
How Is a Differential Diagnosis for Posterior Segment Uveitis Generated?
A differential diagnosis should be developed for each patient with uveitis after completion of a thorough history and ophthalmic examination and before any diagnostic tests are ordered or therapy instituted.9 The exercise of generating the differential diagnosis should be performed even if the diagnosis seems certain clinically. The differential diagnosis is a useful guide for determining which laboratory and ancillary studies should be requested and provides ready diagnostic alternatives if the patient does not respond appropriately to therapy or later develops new ophthalmic or systemic symptoms or signs. A differential diagnosis can be easily developed by considering the answers to the following questions: Is the disease acute or chronic? Is the inflammation granulomatous or nongranulomatous? Where is the inflammation located in the eye? How did the disease respond to previous therapy? Is the disease unilateral or bilateral? What are the demographics of the patient? What associated systemic symptoms and physical signs does the patient have? The significance of the responses to the first four of these questions have been addressed previously in this chapter; therefore, only the last question is considered here.
Although one eye may be affected first, most causes of posterior segment uveitis involve both eyes within the first several months. Causes of uveitis that frequently involve a single eye even after months or years include sarcoidosis, Behçet disease, and infectious causes of uveitis, such as toxoplasmosis, acute retinal necrosis, and postoperative endophthalmitis. Demographic considerations, such as the patient’s age, sex, race, ethnic heritage, geographic residence, dietary habits, animal exposure, travel history, social behaviors, and sexual habits, can lead the ophthalmologist to suspect certain types of uveitis. Age is particularly important because certain causes of uveitis are more common among patients of specific age group. For example, in children younger than 5 years, the most likely cause of posterior segment uveitis is infectious or neoplastic. Children aged 5 years to adolescence with posterior segment uveitis are more likely to have juvenile rheumatoid arthritis with spillover from the anterior segment or intermediate uveitis. Sarcoidosis and primary intraocular lymphoma should be strongly considered in any patient over the age of 50 years with new-onset posterior segment uveitis. The white-dot syndromes tend to affect women more commonly than men. Sarcoidosis is the most frequent cause of uveitis in African Americans in the United States. Patients from ethnic groups characterized by darker skin pigmentation are more susceptible to the VKH syndrome, whereas patients of northern European descent, where the incidence of the HLA-A29 is more common, are more likely to develop birdshot retinochoroidopathy. Lyme disease is most probable in persons living in the northeastern United States, where the vector is commonly found. Patients with acquired toxoplasmosis frequently report a history of exposure to cats or ingestion of undercooked meats. Finally, it must be remembered that patients with high-risk behaviors for human immunodeficiency virus (HIV) transmission are susceptible to the numerous ocular manifestations of acquired immunodeficiency syndrome (AIDS).
Specific systemic complaints and findings on physical examination that may suggest the presence of an underlying systemic disease known to cause uveitis should be sought during the history, with a comprehensive review of systems and, if necessary, consultation with other medical specialists. Symptomatic arthritis and systemic vasculitis in a patient with posterior segment uveitis are not infrequent and may result from numerous systemic diseases, including Behçet disease, sarcoidosis, Lyme disease, IBD, SLE, and other connective tissue diseases that are common to rheumatologist. Neurologic symptoms raise the possibility of MS or Behçet disease, sarcoidosis, the VKH syndrome, and SLE. Headaches and hearing difficulties temporally related to the uveitis suggest the diagnosis of the VKH syndrome and sarcoidosis. Because patients with neurologic symptoms may require neuroimaging and a lumbar puncture, they should be further evaluated in consultation with a neurologist or neuroophthalmologist. Cerebrospinal fluid pleocytosis is a frequent finding in patients with VKH, sarcoidosis, acute posterior multifocal placoid pigment epitheliopathy (APMPPE) and Behçet. Lesions in the brain or optic nerves may be found in patients with sarcoidosis, primary intraocular lymphoma, MS, and Behçet disease. Behçet disease, sarcoidosis, herpes simplex and zoster, syphilis, and Lyme disease are all associated with cutaneous lesions that can be easily differentiated with the help of a dermatologist. The cutaneous lesion erythema nodosum is frequently seen in patients with Behçet disease and sarcoidosis; vitiligo, poliosis, and alopecia are characteristic of the VKH syndrome. Patients should be questioned about the occurrence of oral and genital ulcers, which will raise the possibility of Behçet disease. Diarrhea and abdominal discomfort are common manifestations of IBD. Similarly, cough and shortness of breath are frequent presentations of sarcoidosis. To rule out the possibility of Wegner granulomatosis, sinus complaints should be investigated by an otolaryngologist.
What Are Masquerade Syndromes?
Masquerade syndromes comprise a group of malignant and nonmalignant systemic or primary ocular diseases that clinically present in the eye as an intraocular inflammatory disease or uveitis. In a clinical study, uveitis masquerade syndromes were diagnosed in 5% of patients with uveitis at a tertiary center.10 In patients with the masquerade syndromes, the ocular findings that are presumed to be a result of an immune mediated process in reality are the result of a noninflammatory disease. No specific features can be relied on to distinguish masquerade diseases that can mimic any form of uveitis. In the eye, malignant cells, pigment, pigmented epithelial cells, and melanocytic cells are easily confused with the white blood cells that define a true inflammatory uveitis. Generally, masquerade syndrome should be considered if the patient is younger than 5 or older than 50 years of age at onset of ocular disease, if the clinical features of the ocular disease and course are atypical for the presumed uveitis diagnosis, or if there is no response to adequate immunosuppressive therapy. In some cases, the diagnosis of an ocular masquerade syndrome may be the first manifestation of a potentially life-threatening disease. The most common malignant diseases that masquerade as uveitis are primary CNS or primary intraocular lymphoma in adults and acute leukemias in children. Other malignancies known to masquerade as uveitis include retinoblastoma, choroidal melanomas, metastatic systemic lymphoma, and carcinomas, of which breast and lung are now the most common. Systemic malignancies that metastasize to the eye generally do so late in the disease course and most commonly involve the choroid. Nonmalignant conditions that masquerade as uveitis include hypertension, pigment dispersion, retinal detachment, degenerative retinal diseases, systemic vascular diseases, intraocular foreign bodies, ocular trauma, and hereditary ocular diseases (Table 19–8). Fluorescein angiography is useful in the diagnosis of nonmalignant masquerade syndromes.10 Once the diagnosis of a uveitis masquerade syndrome is entertained, the patient should undergo a thorough medical evaluation including review of systems and imaging, if needed. Diagnostic procedures, including an aqueous paracentesis, vitrectomy, or ocular tissue biopsy, should be performed without delay after the withdrawal of any immunosuppressive therapy, particularly steroids to increase the diagnostic yield. Before the diagnostic procedure, handling of the ocular specimen for routine pathologic studies, such as histology, cytology, immunohistochemistry or electron microscopy, microbiologic culture, serologic assays, or molecular analysis, should be discussed with an ocular pathologist.
| Malignant disorders |
| In adults |
Primary central nervous system or primary intraocular lymphoma |
Systemic non-Hodgkin’s lymphoma metastatic to the eye |
Metastatic carcinoma to the eye |
Breast |
Lung |
Renal |
Uveal melanoma |
| In children |
Leukemia |
Retinoblastoma |
Medulloepithelioma |
Juvenile xanthogranuloma |
| Nonmalignant disorders |
Intraocular foreign body |
Retinal detachment |
Pigment dispersion syndrome |
Hereditary retinal degenerations |
Myopic degeneration |
Hypertension |
Occult ocular trauma |
Ocular ischemic syndrome |
What Are the Clinical Features of Primary Intraocular Lymphoma?
Primary CNS lymphoma (PCNSL) is typically a B-cell extranodal non-Hodgkin lymphoma that can originate in the brain, spinal cord, leptomeninges, and eyes. It represents 4 to 6% of all primary brain tumors and 1 to 2% of all extranodal lymphomas.11 Primary intraocular lymphoma (PIOL) defines a subset of PCNSL in which lymphoma cells are initially present only in the eyes. PCNSL and PIOL are to be distinguished from a systemic lymphoma that has metastasized to the brain and eye, respectively. Although the incidences of both PCNSL and PIOL have increased within the last few decades, both are considered rare malignancies. Studies demonstrating the presence of DNA from the Epstein-Barr virus, human herpes-virus-6 and −8, and, most recently, Toxoplasma gondii, suggest a role for infectious pathogens in the development of PCNSL and PIOL.12 The median age of onset for PCNSL and PIOL is the fifth to sixth decades of life. Because PIOL can masquerade for years as an intermediate uveitis or idiopathic vitreitis, it should be considered high in the differential diagnosis of any patient over 50 years of age with new-onset or persistent uveitis, particularly if there are any neurologic complaints. Of the 60 to 80% of patients with PIOL that eventually develop CNS disease, most do so within 1 to 2 years after the ocular disease is diagnosed.13,14
Floaters and blurred vision are the most common ocular complaints from patients with PIOL. The disease is bilateral in 80% of cases. Anterior segment inflammation in patients with PIOL is typically minimal, although in rare cases a neoplastic hypopyon can occur. The vitreitis in patients with PIOL appears as clumps or sheets of cells that may obscure the retina, which may or may not show yellow punctate lesions, subtle RPE elevations, pigment mottling, or large subretinal lesions that pathologically are lymphoma cells infiltrating between the RPE and Bruch membrane (Fig. 19–2). Atypical findings include iris infiltrates, vascular sheathing, hemorrhagic retinal exudates mimicking a viral retinitis, and the presence of large elevated subretinal mass lesions.15,16 Fluorescein angiography commonly shows punctate hyperfluorescent and hypofluorescent lesions, predominantly within the posterior pole. An important clinical “pearl” is that although the disease initially may respond to immunosuppression, it ultimately becomes resistant to therapy. Another important clinical pearl is that patients with PIOL tend to have a visual acuity much better than would be expected for their degree of vitreitis. This may be because cystoid macular edema, in our experience, is extremely uncommon in eyes with PIOL unless there has been previous ocular surgery.
Evaluation of a patient suspected to have PIOL includes thorough medical and neurologic examinations to rule out evidence of a systemic malignancy, neuroimaging to detect the presence of CNS lesions, and repeated lumbar punctures with cytologic examination of the cerebrospinal fluid to rule out the presence of malignant lymphoid cells. If these examinations are not diagnostic, a vitrectomy should be performed on the eye with the worst vision or most severe vitreitis with immediate cytologic examination of the vitreous specimen to increase the diagnostic yield. If corticosteroids were given as treatment for uveitis, they should be discontinued for at least 1 month before a diagnostic procedure is done. In some cases, a chorioretinal biopsy or fine-needle aspiration biopsy of an elevated subretinal lesion may be required to obtain diagnostic tissue. PIOL is a high grade, large B-cell lymphoma. T-cell phenotypes have been reported but are extremely rare.17 It has been demonstrated that molecular analysis of atypical lymphoid cells for rearrangements of the immunoglobulin heavy-chain gene and measurement of interleukin 10 (IL-10) and IL-6 levels in the cerebrospinal and vitreous specimens are useful adjunctive tools for the diagnosis of PCNSL and PIOL, respectively.18,19 In these specimens, an elevated IL-10 level with an IL-10:IL-6 ratio greater than 1.0 suggests the presence of malignant lymphoid cells.20,21
Present treatment strategies for PCNSL include radiation alone, combined radiation and chemotherapy, and chemotherapy-only regimens.22 The best method of treatment for patients with PIOL or recurrent PCNSL in the eyes only has not been determined. Although ocular radiation is the most commonly reported treatment for PIOL, fewer patients are receiving radiotherapy because of the high incidence of ocular recurrence and the risks for delayed complications, such as radiation retinopathy, optic neuropathy, cataracts, glaucoma, keratoconjunctivitis sicca, persistent corneal epithelial defects, and limbal stem cell loss. Most patients with PIOL are currently being treated with chemotherapy regimens using high-dose methotrexate. The successful use of intravitreal methotrexate for the treatment of PIOL has been described.23,24 The major complication of this therapy is ocular surface disease, which may be treated or prevented with topical folinic acid drops. Despite treatment for PIOL, recurrence is common. Because treatment involves the use of chemotherapy and the high probability of subsequent disease within the brain, patients with PIOL should be managed in consultation with a neurologist or neurooncologist skilled in the treatment of this disease.
What Are the Causes of Posterior Uveitis?
Posterior uveitis defines ocular inflammatory conditions that primarily affect the retina and choroid. As a result of direct damage to these tissues, posterior uveitis and panuveitis are a greater cause of visual morbidity than anterior and intermediate uveitis. Posterior uveitis may be a manifestation of an infectious or non-infectious disease. Common infectious causes of posterior uveitis are parasitic diseases, such as toxoplasmosis and toxocariasis; viral diseases, such as cytomegalovirus (CMV) retinitis, herpes simplex, and varicella zoster viruses, which cause acute retinal necrosis and progressive outer retinal necrosis; fungal diseases, like histoplasmosis, candidiasis, Nocardia asteroides, cryptococcosis; and bacterial diseases, including syphilis, Lyme disease, and tuberculosis. This chapter focuses on the noninfectious causes of posterior uveitis (Table 19–4). Noninfectious causes of posterior uveitis producing focal lesions are uncommon; therefore, solitary retinal or choroidal lesions associated with ocular inflammation should raise the possibility of an infectious disease or an intraocular malignancy. Causes of posterior uveitis characterized by multifocal lesions include multifocal choroiditis, bird-shot retinochoroidopathy, serpiginous choroidopathy, sarcoidosis, and the VKH syndrome. The white-dot syndromes are a group of diseases that cause localized, well-circumscribed inflammatory lesions in the fundus. Posterior uveitides falling under this heading, including the multiple evanescent white-dot syndrome (MEWDS), acute posterior multifocal placoid pigment epitheliopathy (APMPPE), acute zonal occult outer retinopathy (AZOOR), and birdshot retinochoroidopathy, are discussed elsewhere in this text.
What Are the Clinical Features of Multifocal Choroiditis?
Multifocal choroiditis is a primary ocular disease. Clinically, patients with multifocal choroiditis have bilateral ocular findings similar to those of the presumed ocular histoplasmosis syndrome (POHS), including multiple “punched-out” chorioretinal scars ranging 50 to 200 μ at the level of the RPE and choroid and peripapillary pigment changes. Compared with ocular histoplasmosis, the chorioretinal scars in multifocal choroiditis show greater peripheral pigmentation and are more commonly found posterior to the equator (Fig. 19–3). Unlike patients with the ocular histoplasmosis syndrome, who characteristically have a clear ocular media, the posterior segment in patients with multifocal choroiditis typically shows a significant vitreitis that may spill over into the anterior chamber. Optic-disc edema is another frequent finding. Because of the anterior segment inflammation, some researchers have described the disease as multifocal choroiditis with panuveitis.
The diagnosis of multifocal choroiditis is based on the clinical findings and has been made most often in myopic women.31 Visual loss may result from cystoid macular edema, optic neuropathy, and, as in patients with POHS, from subretinal neovascular membrane formation in the macula. Despite its similarities to POHS, a relatively low percentage of tested patients respond to the histoplasmin skin test. The cause of the disease is unknown. Treatment involves the use of corticosteroids and other immunosuppressive agents. Laser photocoagulation may be considered for subretinal neovascularization unresponsive to medical therapy. Because multifocal chorioretinal lesions are a feature of many forms of both infectious and noninfectious posterior or panuveitis, multifocal choroiditis must be differentiated from entities in the white-dot syndrome, sarcoidosis, birdshot retinochoroidopathy, PIOL, VKH syndrome, and POHS. The diagnosis probably should be excluded if there is evidence of any systemic disease known to be associated with uveitis.
What Are the Clinical Features of Serpiginous Choroidopathy?
Serpiginous choroidopathy is a rare condition that accounts for 1 to 2% of all posterior intraocular inflammatory disorders.32 It is a clinically defined primary ocular disease characterized by progressive destruction of the RPE and choriocapillaris with secondary involvement of the outer retina. Serpiginous choroidopathy has been described in the literature under a variety of names, including helicoid peripapillary chorioretinal degeneration, geographic choroidopathy, macular geographic helicoid choroidopathy and, most recently, relentless placoid chorioretinitis.33 Serpiginous choroidopathy is typically diagnosed in the fourth to sixth decades of life, with most patients complaining of blurred vision, metamorphopsia, photopsia, and central or paracentral scotomas. It is typically a bilateral disease, although the presentation and course may be asymmetric. Examination of the anterior segment rarely shows evidence of inflammation. The vitreous is also typically quiet but may show a mild vitreitis (grade 1+ or less) in 25 to 50% of affected patients.34 Diagnosis is made by the characteristic clinical picture of yellow–white serpiginous or pseudopodial chorioretinal lesions spreading centrifugally from the optic disc temporally more so than nasally (Fig. 19–4). Active, regressing, and inactive components of the disease are often present concurrently in different areas of the expanding lesions. Active areas, most frequently seen along the edges of the lesion, are recognized by the appearance of grayish white elevations at the level of the RPE. The inactive areas are characterized by atrophy of the choriocapillaris and overlying outer retina and marked degrees of RPE hyperpigmentation. Infrequently, cystoid macular edema, vascular sheathing, RPE detachments, disc neovascularization, and subretinal neovascularization may occur, with the last causing severe visual loss.