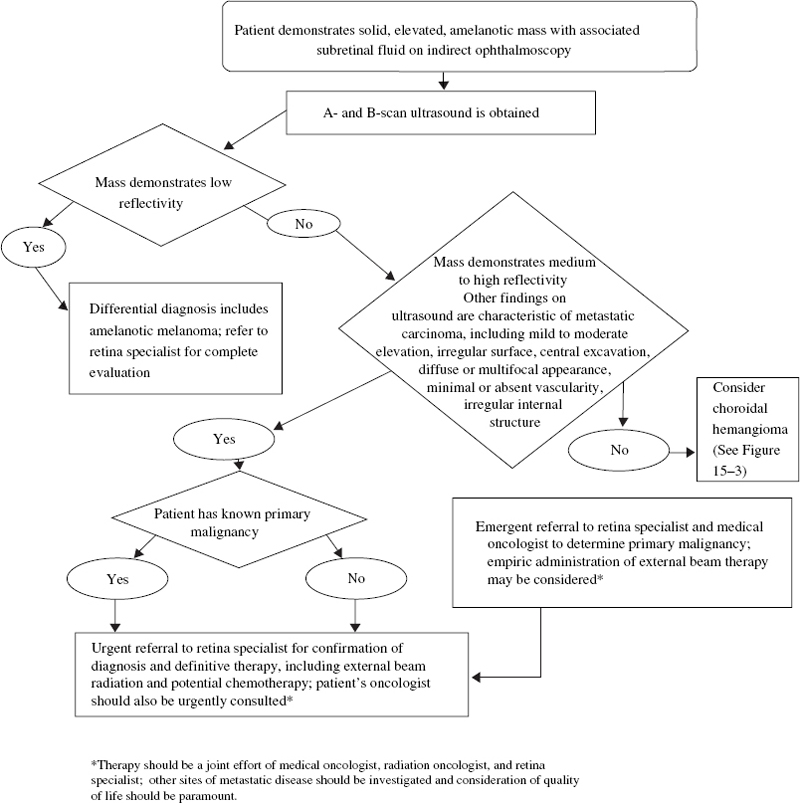
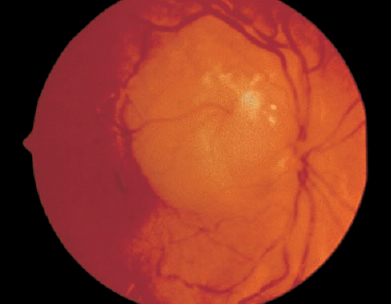
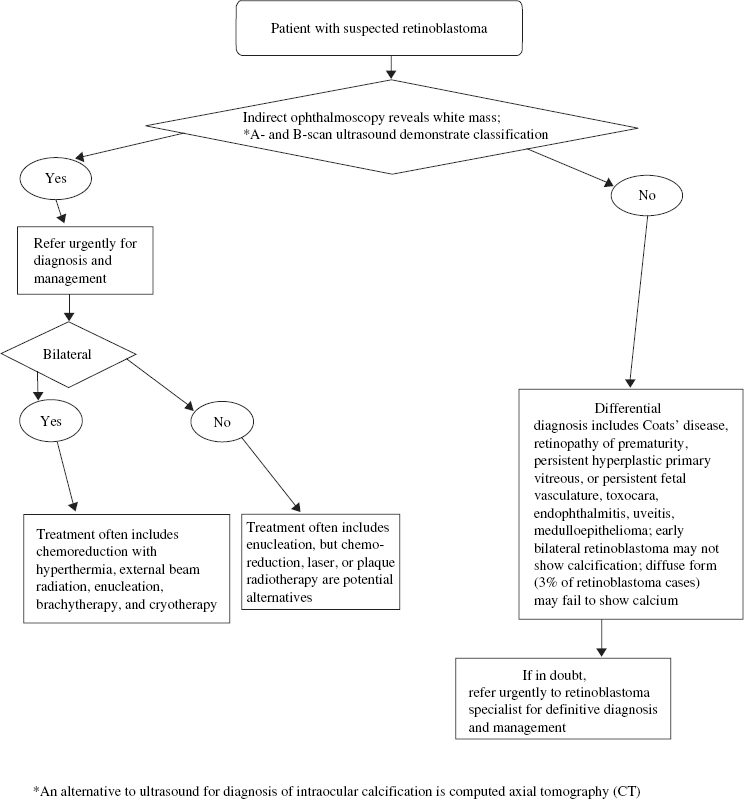
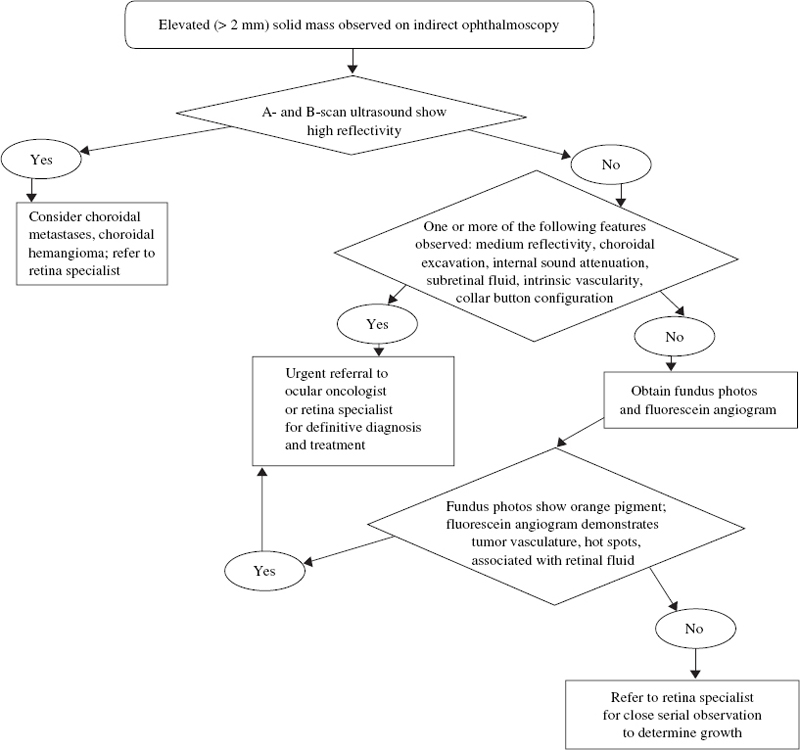
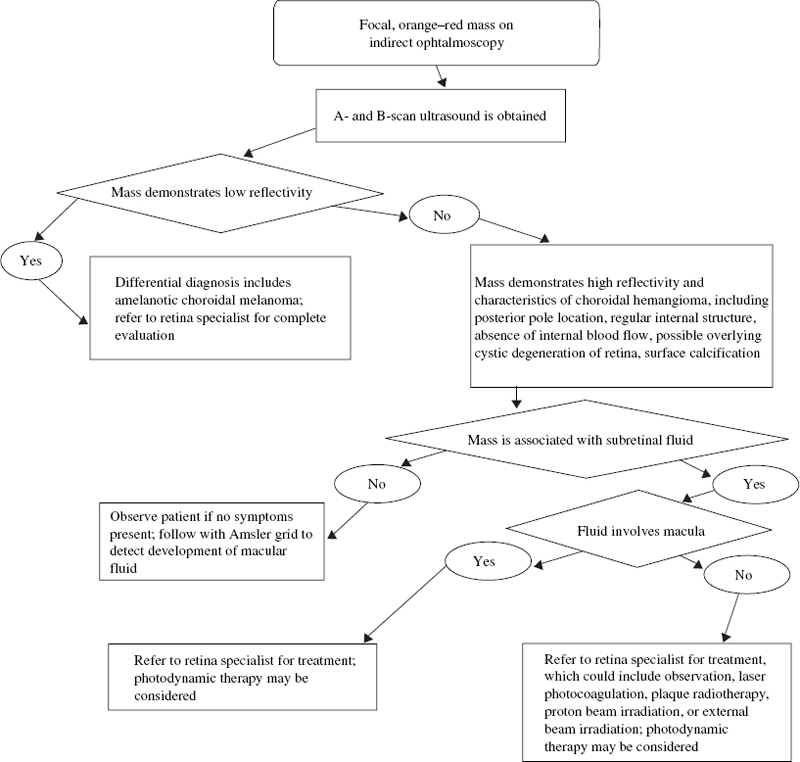
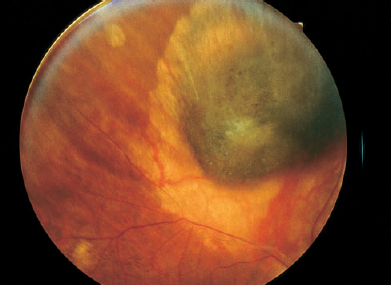
SECTION III Retinoblastoma is the most frequent intraocular tumor in children.1,2 It affects between 1 in 15,000 to 1 in 20,000 live births annually, resulting in 200 to 300 children in the United States being affected by the disease.2,3 Retinoblastoma accounts for 12% of infant cancers.3 There is no sexual or racial predilection for the development of this disease.3 There is a higher incidence of retinoblastoma in the developing world.4–6 Uveal melanoma is uncommon in black populations; thus, retinoblastoma is the most common intraocular tumor in Africa.6 The retinoblastoma gene occupies 200 kilobases on chromosome 13 in region 13q14.7 The product of the retinoblastoma gene acts as a tumor suppressor and plays an important role in regulating cell growth and differentiation.8 Both alleles must be mutated at the RB1 locus for retinoblastoma to develop.7–11 These two genetic mutations correlate with the “two-hit hypothesis” proposed by Knudson.12 In 1971, Knudson evaluated epidemiologic data in 48 cases and concluded that retinoblastoma likely results from either a germinal or somatic mutation. He hypothesized that in heritable retinoblastoma the first “hit” occurs in the germline and is inherited or develops spontaneously in all cells of the body. The second “hit” is a somatic mutation that develops within retinal cells. In patients with sporadic unilateral retinoblastoma, both hits are somatic mutations within a single retinal cell, and there is normal chromosomal composition elsewhere in the body.12 Subsequent to Knudson’s hypothesis, Comings proposed that mutations responsible for the phenotype could inactivate both alleles of a single responsible gene.13 The cloning of the retinoblastoma gene and subsequent detailed genetic analysis confirmed the validity of these scientific proposals.7–14 Approximately one third of children affected by retinoblastoma will have bilateral disease. Bilateral retinoblastoma results from a germline mutation at the retinoblastoma locus or mosaicism affecting primordial cells in the retina. Bilateral disease is associated with an underlying mutation that is transmissible in the germline to offspring. In these patients there is a risk for midline brain tumors, primitive neuroectodermal tumors, and second malignancies such as osteogenic or soft-tissue sarcomas. Unilateral retinoblastoma usually develops due to a mutation occurring somatically within a single retinal cell at the retinoblastoma locus. Although most unilateral patients have sporadic, somatic mutations, up to 19% of children who present with unilateral disease have an underlying germline retinoblastoma mutation. These patients are also at risk for second tumors and midline brain tumors.15,16 The retinoblastoma gene was the first tumor suppressor gene to be identified. Deletion or mutation in a tumor suppressor gene results in deregulated cell growth and proliferation. Children with germline mutations at the retinoblastoma locus demonstrate a life-long tumor predisposition. Treatment of the primary ocular tumors in these children can increase this already elevated risk of second tumor development.17–20 Therapy aims for cure primary disease while minimizing the risk for second tumors. On light microscopic examination, viable retinoblastoma cells are basophilic, whereas necrotic retinoblastoma tumors are eosinophilic. In areas of necrosis, foci of calcification are typically seen. Another characteristic finding is the presence of basophilic material deposited in the walls of blood vessels; this represents precipitated DNA and chromatin. The tumor is composed of neuroblastic cells that can develop from any nucleated retinal layer. Poorly differentiated tumors are composed of small, round cells with a high nuclear-to-cytoplasmic ratio and hyperchromatic nuclei. More differentiated tumors contain rosettes or florets. The Homer–Wright rosette consists of radially arranged cells surrounding a central tangle of neurofilaments. These rosettes are observed in tumors such as neuroblastoma, medulloepithelioma, sinonasal undifferentiated carcinoma, and medulloblastoma. The Flexner–Wintersteiner rosette is composed of cells arranged in a circular fashion around a central empty lumen. An internal limiting membrane separates the cells’ borders from the central lumen. Retinocytoma and the most well-differentiated forms of retinoblastoma contain florets composed of benign-appearing tumor cells demonstrating photoreceptor differentiation. Pear-shaped eosinophilic processes extend along one side of the tumor cells; these cells are bound together by terminal bars that represent the outer limiting membrane of the retina.21 Retinoblastoma occurs very early in life. Germline retinoblastoma mutations likely develop prenatally. Patients with a known family history of retinoblastoma should be examined shortly after birth and followed carefully thereafter. All siblings and offspring of retinoblastoma patients should be evaluated at the earliest possible opportunity and followed with serial examinations under anesthesia (EUA). FIGURE 15–1. Untreated peripapillary retinoblastoma. Lesion demonstrates the pinkish white gelatinous appearance typical of active tumor. Bright white flecks represent intrinsic tumor calcification. Patients with small tumors and no family history may present with strabismus or decreased visual acuity, depending on the location of the tumor. Pediatric patients should receive full retinal evaluations to exclude retinoblastoma or other retinal pathology as the etiology of their strabismus. Such complete evaluations are important because early diagnosis and treatment may spare the patient systemic toxicity associated with radiation or chemotherapy. Early retinoblastomas are round, slightly elevated, gray–white intraretinal masses. These tumors frequently demonstrate an intrinsic vasculature that appears as a fine network of pink vessels within the main tumor mass. Flecks of intrinsic calcification may be present within larger tumors, appearing as dense, white opacifications (Fig. 15–1). Substantial subretinal fluid may be associated with active retinoblastoma. Larger tumors may seed the subretinal or vitreous space, and tend to present as unilateral or bilateral leukokoria. The retrolental tumor mass may enlarge in an endophytic pattern toward the vitreous cavity or in an exophytic pattern toward the choroid and sclera. A diffuse infiltrating pattern is less common but more likely to be misdiagnosed. Retinoblastoma rarely grows diffusely within the retina without producing a discrete mass. Patients who present with presumed endophthalmitis resulting from the appearance of pseudohypopyon are likely to have the diffuse form of retinoblastoma.22–24 Neovascularization of the iris occasionally occurs in retinoblastoma associated with posterior segment ischemia,25 which can lead to neovascular glaucoma and spontaneous hyphema without a history of trauma. Patients with endophytic tumors may have vitreous seeding; seeds may also be shed into the anterior chamber, giving a clinical appearance similar to iritis. The tumor may infiltrate the iris, producing heterochromia iridis. Preseptal cellulitis is an uncommon manifestation of retinoblastoma and is associated with secondary periocular inflammation. In the developing world, retinoblastoma frequently presents with corneal perforation, buphthalmos, proptosis due to posterior orbital extension, or a prolapsing orbital mass.26 The initial evaluation of suspected retinoblastoma requires a detailed medical history. The prenatal history should address any maternal history of rubella infection or other prenatal exposures and any medical complications that arose during pregnancy. Eliciting a perinatal history of prematurity, oxygen therapy, and labor and delivery complications is helpful in excluding retinopathy of prematurity in the differential diagnosis. History of pica and contact with puppies or cats should be elicited to evaluate the possibility of toxocariasis. The child’s physical and mental development should be assessed. A detailed family history should be taken and a detailed family tree recorded. Family history of retinoblastoma, blindness in any generation, eye loss in family members, and a history of osteosarcoma or other cancer should be noted. A review of systems should focus on symptoms of weight loss, cachexia, failure to thrive, or other manifestations of systemic malignancy. Physical examination initially should focus on the child’s general appearance and behavior. Dysmorphic features and developmental delay suggest 13q minus syndrome, which occurs in association with retinoblastoma in a small percentage of patients.27 Visual acuity should be assessed in each eye. A slit-lamp examination and indirect ophthalmoscopy may narrow the differential diagnosis and determine which additional tests are necessary. Definitive examination is performed under anesthesia. The EUA should include external eye examination, intraocular pressure recording, and anterior segment evaluation with a portable slit lamp or under the operating room microscope. Indirect ophthalmoscopy with scleral depression and fundus photography should be performed. Simulating lesions, such as toxocariasis, Coats’ disease, or retinopathy of prematurity (ROP), can usually be distinguished through clinical evaluation. Ultrasonography should be performed to narrow the differential diagnosis. Retinoblastoma is suggested on A-scan by highly reflective internal echoes consistent with intrinsic tumor calcification. Rapid attenuation of the normal orbital pattern presents as dampening of echoes posteriorly. B-scan ultrasonography characteristically demonstrates a dome-shaped mass within the vitreous space. A low-gain setting is useful to demonstrate highly reflective echoes corresponding to calcium within this intraocular mass. Calcification within the tumor produces attenuation and reflection of the sound wave, resulting in soft-tissue shadowing in the orbit. Computed axial tomography (CT) scan of the orbit should be obtained in all patients with confirmed retinoblastoma on EUA. This study confirms intrinsic tumor calcification and delineates tumor extension into the orbit. Magnetic resonance imaging (MRI) is the most sensitive study to evaluate for pinealoblastoma, optic nerve disease, or intracranial extension. Retinoblastoma may be diagnosed clinically. Invasive intraocular techniques such as fine needle aspiration are never indicated because they produce a high associated risk for retinoblastoma dissemination.28 Any child who presents with retinal detachment, hyphema, or other conditions which preclude view of the posterior pole, should have retinoblastoma excluded by imaging studies (ultrasound, CT, or MRI) before any surgical procedure is performed. Retinoblastoma should always be considered carefully in the differential diagnosis of pediatric ocular disease. The most frequent treatment for retinoblastoma is enucleation; this procedure is required in 90 to 95% of unilateral patients (Fig. 15–2). Chemoreduction with application of local therapies, such as cryotherapy, laser ablation, or plaque radiotherapy, provides an alternative to enucleation in some patients. External-beam radiation therapy is highly effective in curing primary disease but may be associated with disfiguring midface hypoplasia and an increased risk of a second tumor development.17 The choice of therapy must consider the visual potential of each eye, the age of the patient, the stage of disease at presentation, and the location of tumors. Patients with asymmetric disease, with one eye having greater than 50% of ocular volume occupied by the tumor, may benefit from enucleation of the more involved worse eye and local therapy applied to the better eye. In relatively symmetric disease, treatment such as external-beam radiation or chemoreduction should be considered. Patients with retinoblastoma may have recurrent tumors throughout the first years of their lives. Any systemic therapy has the potential to increase the bilateral retinoblastoma patient’s already elevated predisposition for a second tumor. Patients should be followed prospectively for second tumor development by knowledgeable pediatric oncologists. Retinocytoma or retinoma are spontaneously regressed retinoblastomas. These lesions have the clinical appearance of treated retinoblastoma. In a series of 920 consecutive retinoblastoma patients, the incidence of retinocytoma was estimated at 1.8%.29 Others have estimated this incidence to be as high as 10% among all retinoblastoma patients.30 On average, retinocytoma presents at a later age than retinoblastoma. The median age at diagnosis for retinocytoma is 15 years, with a range from 4 to 45 years.29 A small number of retinocytomas have been evaluated histopathologically; most of these tumors are clinically observed. Eagle described the histopathologic appearance of a retinocytoma followed for 3 years with no clinical change until a rapid growth phase in the third year indicated enucleation. Histopathology revealed a poorly differentiated retinoblastoma within the region of rapid growth. The remainder of the tumor demonstrated features more characteristic of retinocytoma, including bland nuclear morphology, a complete absence of mitoses, fibrillar eosinophilic stroma, occasional calcific foci, and dispersed florets. Immunohistochemical analysis confirmed that positivity for S-100 protein, glial fibrillary acidic protein, and retinal S-antigen was confined to the more differentiated, basal portion of the tumor.31 Other histopathologic reports describe retinocytomas as tumors composed of normal photoreceptor cells, bipolar cells, Muller cells, and astroglia.32 FIGURE 15–2. Clinical pathway: Evaluation of suspected retinoblastoma. Spontaneously regressed retinoblastoma also has been described in phthisical eyes.32–34 In these cases, the tumors display coagulative necrosis and dense calcification. Only the outlines of fossilized tumor cells are visible by light microscopy. Secondary hyperplasia of the retinal pigment epithelium, glial cells, and ciliary epithelium may be observed.33 Retinocytomas are frequently asymptomatic and diagnosed by routine ophthalmologic examination. These tumors may be discovered in asymptomatic family members of retinoblastoma patients. Retinocytomas resemble treated retinoblastoma in that they are frequently small tumors with intrinsic calcification. The tumors may be slightly elevated, gray, homogeneous, and translucent.34 A surrounding halo of chorioretinal atrophy may be observed, suggesting previous tumor regression.29 Malignant degeneration is possible in retinocytomas. The diagnostic approach parallels that of retinoblastoma. A complete medical history, family history, and physical examination with indirect ophthalmoscopy and imaging studies should be undertaken. Fundus photos and A and B-scan ultrasound should be obtained serially to document stability. Years of observation may be required to distinguish retinoblastoma from retinocytoma. A series by Singh and colleagues suggested that the risk for malignant transformation in retinocytomas is about 4%.29 Thus, patients with a presumed diagnosis of retinocytoma require close serial observation. Fundus photography is useful in documenting tumor stability. Ultrasound evaluation of basal dimensions and tumor thickness should be obtained, and repeat indirect ophthalmoscopic examinations should evaluate the tumor for any signs of activity. A presumed retinocytoma with documented growth in basal dimension, increase in thickness, development of associated fluid, or signs of seeding should be treated as a retinoblastoma.31 Melanocytoma is a nevus usually observed in the region of the optic disc but also observed elsewhere in the choroid. These congenital, nonhereditary lesions are composed of proliferations of pigmented melanocytes. Over time, the tumors generally remain stationary in clinical appearance and demonstrate very limited malignant potential. Melanocytoma occasionally is confused with choroidal melanoma or choroidal nevi.35 The incidence and prevalence of melanocytoma are not precisely known. Although these tumors are most likely present at birth, the age of diagnosis is variable, ranging from 14 to 80 years. Melanocytomas are usually unilateral. The incidence in women is slightly higher than the incidence in men, with 58% of cases reported in females. Around 50% of melanocytomas occur in patients of African heritage, in contrast to melanoma, which occurs in fewer than 1% of African-American patients.35–37 On light microscopy, melanocytoma is composed of plump polyhedral cells with bland nuclei. Melanocytoma cells are densely pigmented, and bleaching is required to visualize cellular details. Cells have an oval to round shape with abundant cytoplasm, small nuclei, and few nucleoli. These tumors are frequently composed of two cell types: Type I cells are large, round, and deeply pigmented, with large melanosomes, and type II cells are smaller, spindle-shaped, and less pigmented, with small melanosomes. Type II cells are less metabolically active and have a slower pattern of growth, and cystic spaces may be observed within the main tumor mass. There is no chromatin margination, and mitotic figures are not observed. These tumors may be associated with overlying drusen and attenuation or hyperplasia of the retinal pigment epithelium (RPE).38 Melanocytomas of the optic-nerve head are generally asymptomatic.25 These tumors may enlarge slowly and infiltrate locally, resulting in afferent pupillary defects or visual fields defects.39 The melanocytic cells that constitute these tumors can produce compression of the axons of the optic nerve with optic disc edema and occasional progressive vision loss.39 Retinal vascular obstruction is not frequently observed in association with melanocytoma but has been associated with the development of severe neovascular glaucoma.40 On indirect ophthalmoscopy, melanocytomas present clinically as elevated, dark brown to black masses. They frequently show a fibrillated tumor margin that recapitulates the peripapillary nerve-fiber layer of the retina. These pigmented tumors may be eccentric, straddling one edge of the optic nerve, or they may involve the entire surface of the optic-nerve head. Other clinical features include overlying RPE changes, drusen, and sheathing of retinal vessels. The optic disc may appear anomalous in configuration or may have an edematous appearance. Melanocytomas also occur in the iris, ciliary body, and choroid. These deeply pigmented tumor cells generally produce sharply circumscribed masses.25 A diffuse growth pattern with involvement of the majority of the uveal tract has also been observed.41 The indirect ophthalmoscopic features of melanocytoma are frequently diagnostic. Serial fundus photos are indicated to monitor these tumors for growth. Visual field changes associated with melanocytoma of the optic disc include enlargement of the blind spot and nasal step and are related to absolute nerve-fiber bundle defects. Visual fields may be monitored to confirm clinical stability.39 Progressive growth or progressive loss of vision suggests malignant transformation. Fluorescein angiographic studies of melanocytoma demonstrate hypofluorescence throughout all phases of the angiogram; this is associated with blockage by the intense pigmentation of densely packed melanocytoma cells, with little associated vascularity. Hyperfluorescence of the disc may be associated with optic nerve edema. Ultrasonography may be helpful in distinguishing melanocytoma from choroidal melanoma. Melanocytomas are characteristically smooth, dome-shaped, minimally elevated lesions that are highly reflective, with regular internal structure and minimal internal vascularity.42 Melanocytomas may progress to malignant transformation into malignant melanoma, although such transformation is infrequent. Extrapapillary melanocytoma is more likely to undergo malignant transformation than a tumor that overlies the optic-nerve head. In juxtapapillary melanocytoma, the choroidal component is more likely to undergo malignant transformation.25 Melanocytoma is managed with close observation and careful follow-up. If progressive growth and visual loss are clearly documented, treatments appropriate for uveal melanoma should be considered. A melanocytoma is normally managed in the same manner as a choroidal nevus, with observation and serial fundus photography. In cases of documented growth and suspected malignant transformation, all tests used in the diagnosis of uveal melanoma are indicated. Fluorescein angiographic (FA) features and ultrasound characteristics will frequently demonstrate a melanoma pattern; this is helpful in forming an accurate diagnosis and in choosing the appropriate therapy. Choroidal melanoma is the most common primary intraocular malignancy in adults; it is extremely rare in children. The incidence of choroidal melanoma in the United States is six cases per one million population43,44; it increases with age, from about 2.5 per million between the ages of 15 and 44 to 25 per million after the age of 65.43–46 The diagnosis is more frequent in white populations.43,44 Asian and African-American populations are rarely affected.43–46 Choroidal melanoma characteristically produces a dome-shaped subretinal mass with variable pigmentation ranging from mostly amelanotic to heavily pigmented. Clumps of orange pigmentation at the level of the RPE represent lipofuscin. Localized serous retinal detachment is frequently observed.47 The size of the tumor in largest basal dimension and the predominant cell type are the two most important pathologic variables in the prognosis.47 Uveal melanomas are classified according to the modified Callender classification as predominantly spindle cell type, epithelioid, or mixed cell type.48 Spindle-cell tumors can be classified as spindle-A or spindle-B. Spindle-A cells have low nuclear-to-cytoplasmic ratio. In these cells, nucleoli are absent, intranuclear vacuoles may be observed, and a chromatin strip may extend centrally through the nucleus. Tumors composed entirely of spindle-A cells are considered to be nevi. Spindle-B cells have higher nuclear-to-cytoplasmic ratio, open chromatin, and more prominent nucleoli. Tumors composed of a mixture of spindle-A and spindle-B cells are classified as spindle cell melanomas. Epithelioid melanoma cells resemble epithelium, with abundant cytoplasm and large oval to round nuclei. These cells have large nucleoli and very coarse chromatin. A tumor is epithelioid according to the Callender classification if it is composed of more than 50% epithelioid cells. Tumors composed of less than 50% epithelioid cells are mixed cell tumors.48 Choroidal melanoma located in the posterior pole may present with decreased vision. Flashes, floaters, or photopsias may narrow the differential diagnosis, especially when there is associated exudative detachment or hemorrhage caused by rupture of the tumor through Bruch’s membrane. There may be a scotoma corresponding to the tumor or visual field changes associated with exudative detachment. Large or anterior tumors may produce angle-closure glaucoma and pain as a result of elevated intraocular pressure. Rarely, choroidal melanoma may present with orbital discomfort associated with scleritis.49 Frequently, choroidal melanomas are completely asymptomatic and diagnosed on routine ophthalmologic examination.25 Choroidal melanoma is frequently diagnosed through indirect data because biopsy is rarely required. Intraocular biopsy can be external through fine-needle aspiration or internal by a vitrectomy with a biopsy approach. Careful application of indirect methods, including indirect ophthalmoscopy, fundus photography, FA, and A and B-scan echography assures an accurate diagnosis (Fig. 15–3). The Collaborative Ocular Melanoma Study (COMS) reported a 99.7% accuracy of diagnosis following indirect diagnostic testing in 1532 cases.50,51 Disciform scar, large choroidal nevi, choroidal metastases, choroidal hemangioma, choroidal detachment, granuloma, and melanocytoma can simulate choroidal melanoma. Appropriate testing is necessary to narrow the differential diagnosis (Fig. 15–4). On indirect ophthalmoscopy, choroidal melanoma is characteristically elevated, dome-shaped, pigmented (although amelanotic melanoma can show minimal pigmentation), and associated with subretinal fluid. Orange pigmentation may be associated with the main tumor mass. Tumors that have broken through the Bruch’s membrane display a characteristic, although not pathognomonic, collar-button configuration.52 These tumors may be associated with overlying vitreous or subretinal hemorrhage. Retinal invasion may be velvety brown and adjacent to the primary mass. Infrequently, choroidal melanoma may demonstrate a diffuse pattern; these forms tend to be large in basal dimension and relatively thin. Diffuse tumors can extend extraocularly.53–55 Fundus photographs may demonstrate the clinical features and establish a baseline for serial comparison. FA frequently demonstrates “hot spots” or point sources of leakage. An intrinsic tumor vasculature may be observed56,57 and may be particularly evident in the apical collar button area. FA is also useful in the diagnosis of similar lesions, such as eccentric disciform scar, arterial macroaneurysm, choroidal hemangioma, or choroidal detachment. A and B-scan ultrasound studies are particularly useful in narrowing the differential diagnosis. Compared with other intraocular masses, choroidal melanoma demonstrates characteristically low to moderate reflectivity on A-scan evaluation. B-scan demonstrates choroidal excavation, internal sound attenuation, and the presence of intrinsic vascularity. A break through Bruch’s membrane with a characteristic collar button configuration may be confirmed by echography.42 A careful history and physical examination are important in the initial evaluation. Questions should be directed to the patient’s state of overall health. Recent weight loss, cachexia, malaise, abdominal discomfort, breathing difficulties, or shortness of breath should be explored during the review of symptoms. Initial history should also focus on the patient’s visual symptoms and should explore other causes of decreased vision such as glaucoma, diabetic retinopathy, or age-related macular degeneration. Chemotherapy for treatment of a previous malignancy should be discussed because this exposure may aggravate radiation-associated retinopathy. The patient’s occupational and recreational visual needs must be addressed because these could impact on the chosen therapy. It is particularly important to assess the visual potential of both the involved and uninvolved eye. Treatment of primary intraocular melanoma is not indicated if the patient has already developed systemic metastases. Although choroidal melanoma patients usually do not present with metastases, these should be excluded by liver function tests and chest radiography. If liver function tests are elevated, an imaging study of the liver, including ultrasound, MRI, or CT imaging should be obtained. Less common sites of metastasis are the lungs, skin, bones, and brain.58 Once metastatic disease is eliminated as a possibility, the patient’s treatment may be determined. Accurate diagnosis and treatment of choroidal melanoma require informed consent from the patient with knowledge of all potential treatments and available outcomes data.50,51,53,59–67 Enucleation is a standard therapy in the treatment of large and medium-sized choroidal melanomas. The COMS compared preenucleation external beam radiation followed by enucleation to enucleation alone for large uveal melanoma.50,51,59 There was no statistically significant difference in five year survival.60 Enucleation is the best treatment option for eyes with very large choroidal melanomas or with very limited visual potential. An ocular implant is usually placed at the time of enucleation. FIGURE 15–3. Clinical pathway: Evaluation of suspected choroidal melanoma. Radioactive plaques may be applied to administer high-dose radiotherapy (brachytherapy). A variety of isotopes have been used effectively, including ruthenium 106, iridium 192, iodine 125, and cobalt 60.25 Iodine 125 is the most frequently used isotope in the United States. Complications of brachytherapy are dose dependent. Vision loss is generally more pronounced for tumors involving the macula or optic nerve. Late complications can include optic neuropathy and radiation retinopathy. During brachytherapy, the maximum dose of radiation is delivered in the region contiguous to the plaque; thicker tumors require more radiation delivered to the tumor base to assure that the tumor apex receives adequate irradiation.25 The COMS Medium Tumor Trial demonstrated that radiation delivered by plaque radiotherapy (Fig. 15–5) and enucleation are equally effective in controlling primary disease at up to 12 years of follow-up.61,63 For this treatment, tantalum marker clips are surgically placed at the margins of an uveal melanoma before radiation, providing for beam localization.68 High-energy radiation is delivered with charged particles (protons or helium ions) in an accelerated linear beam. The energy is delivered in a homogeneous fashion; there is no gradient in dose between tumor apex and base. The lateral spread of radiation from the charged particle beam is considerably less than from brachytherapy; the dose of radiation drops from 100 to 0%, 2.3 mm from the beam edge. This method may be advantageous in tumors that have a peripapillary or macular location. Charged particle radiation, like brachytherapy, may result in radiation-associated retinopathy or optic neuropathy.69,70 Although this method of radiation delivery has not been assessed in prospective, multicenter, randomized trials, it has been used for many years and is considered as effective as radiation delivery by brachytherapy.71–82 FIGURE 15–4. Clinical pathway: Evaluation of circumscribed choroidal hemangioma. Small trials have reported efficacy for transpupillary thermal therapy delivered by diode laser.67,83–93 Follow-up for this relatively new mode of therapy is short, and large, prospective, randomized trials have not compared this treatment with more established methods of treatment such as enucleation or radiation. Diode therapy may be indicated for tumors that threaten vision in uniocular patients. This method may also reduce the thickness of the tumor, allowing a reduction in radiation dose following the application of heat.67,83–93 Until long-term clinical outcomes data are available, this treatment must be considered investigational.8 FIGURE 15–5. Treated choroidal melanoma. Tumor exhibits a slate gray regression pattern with surrounding choroidal atrophy. Eye-wall resection and endoresection have been recommended for iris or ciliary body melanoma and occasionally in patients with choroidal melanoma. The outcomes from eye-wall resection have been reported in single center case series only. The technique is difficult, and the tumor often is not eradicated in the operative margins.94–97 Choroidal metastases occur most often as manifestations of breast cancer in women and lung cancer in men. Carcinomas are the most frequently observed ocular metastases, whereas sarcoma and melanoma are less frequent. Carcinoma of the gastrointestinal tract, kidney, thyroid gland, pancreas, and prostate occur with even lower frequency. In published series, 74 to 94% of uveal metastases involve the choroid.98–100 The incidence of choroidal metastases has increased with the prolongation of survival in cancer patients. Choroidal metastases represent the most frequent malignancy observed in the eye.98 Over one third of breast cancer patients have choroidal metastases.101 Metastases to the choroid are observed most frequently in middle-aged or older patients, and, with the exception of metastatic choroidal disease in leukemia patients, choroidal metastases are rare in children. Choroidal metastases diffusely involve the choroid, are frequently multifocal, and are associated with exudative retinal detachment. The histopathologic appearance of metastases often corresponds to the primary malignancy, particularly when the metastatic lesion is well differentiated. Metastatic carcinoma of the breast may demonstrate glandular structures, whereas metastatic oat cell carcinoma may show an alveolar pattern. Metastatic tumors can be less differentiated and may not resemble the primary tumor; in these cases, special stains and ultrastructural analysis may be informative.21 Choroidal metastases that involve the posterior pole present with decreased vision. These lesions are characteristically creamy yellow at the level of the choroid. Metastatic tumors may be multiple and frequently are associated with substantial subretinal fluid. Choroidal metastatic tumors can grow dramatically over time; thus, early treatment should be undertaken to maintain vision. These tumors frequently produce reactive RPE changes referred to as peau d’orange. When retinal detachment is extensive and bullous, subretinal fluid may shift dramatically. Large metastatic tumors, particularly those involving the anterior uvea, may result in secondary glaucoma. Patients with suspected choroidal metastases require an immediate medical history and physical examination. Urgent referral to a medical oncologist is indicated. If the primary tumor is unknown, then appropriate medical evaluation, imaging studies, and appropriate tissue biopsy should be undertaken. The metastatic choroidal lesion should generally be treated with external-beam radiation therapy. Fluorescein angiography reveals initial hypofluorescence in the laminar venous phase with late hyperfluoresence of the mass.102 Echographically, metastatic choroidal tumors usually show moderate elevation, although high elevation occasionally is observed (Fig. 15–6). Differentiation between suprachoroidal effusion or hemorrhage should be made (Fig. 15–7). Multifocality, an irregular anterior surface and central excavation are frequently observed. B-scan demonstrates a diffuse, subclinical thickening of the choroid. A-scan demonstrates moderate to high reflectivity with an irregular internal structure.42 Choroidal metastases require urgent imaging of the brain and orbits to exclude concomitant central nervous system (CNS) metastases; these must be considered in planning ports for subsequent radiation therapy.
Intraocular Mass, or Lesions Associated with an Elevated Retinal Surface
15
Intraocular Mass
Retinoblastoma
What Are the Incidence and Epidemiology of Retinoblastoma?
What Are the Underlying Genetics of Retinoblastoma?
What Are the Pathologic Features of Retinoblastoma?
How Does Retinoblastoma Present Clinically?
What Is the Appropriate Diagnostic Approach for Retinoblastoma?
How Is Retinoblastoma Treated?
Retinocytoma
What Are the Epidemiology and Incidence of Retinocytoma?
What Are the Pathologic Features of Retinocytoma?
How Does Retinocytoma Present Clinically?
What Is the Appropriate Diagnostic Approach for Retinocytoma?
How Is Retinocytoma Treated?
Melanocytoma
What Are the Epidemiology and Incidence of Melanocytoma?
What Are the Pathologic Features of Melanocytoma?
How Does Melanocytoma Present Clinically?
What Is the Appropriate Diagnostic Approach for Melanocytoma?
How Is Melanocytoma Treated?
Choroidal Melanoma
What Are the Epidemiology and Incidence of Choroidal Melanoma?
What Are the Pathologic Features of Choroidal Melanoma?
How Does Choroidal Melanoma Present Clinically?
What Is the Appropriate Diagnostic Approach for Choroidal Melanoma?
How Is Choroidal Melanoma Treated?
ENUCLEATION
RADIOACTIVE PLAQUE (BRACHYTHERAPY)
CHARGED PARTICLE RADIATION
TRANSPUPILLARY THERMAL THERAPY
EYE-WALL RESECTION
Choroidal Metastases
What Are the Epidemiology and Incidence of Choroidal Metastases?
What Are the Pathologic Features of Choroidal Metastases?
How Do Choroidal Metastases Present Clinically?
What Is the Appropriate Diagnostic Approach for Choroidal Metastases?
Ento Key
Fastest Otolaryngology & Ophthalmology Insight Engine