Fig. 19.1
Cumulative incidence of second malignancy following diagnosis of retinoblastoma in patients with hereditary and nonhereditary retinoblastoma (a) and in the hereditary retinoblastoma with and without radiation treatment (b) (Data derived from Wong et al. [8])
19.2.2 Effects of Radiation Therapy
It is unknown which factors definitively predispose patients to developing second malignancies, and there are obviously many factors yet to be understood. However, in addition to increasing the incidence of non-ocular tumors, radiation therapy appears to influence the age of onset, location, and type of non-ocular cancer (Fig. 19.1b). For many years, it was assumed that second non-ocular tumors in heritable retinoblastoma patients were a direct consequence of radiation dosing. However, when lower doses of radiation were employed, second malignancies continued to occur. This is discussed further in Chap. 14.
19.2.2.1 Three Subsets of Patients
Further study of these patients who continued to get second non-ocular tumors after dose reduction revealed three distinct subsets of patients. The first were those who had received radiation to the orbits but developed second malignancies remote from the radiation field. The second subset of patients developed second tumors in the head and neck area mimicking radiation-induced malignancies but had never received radiation therapy. Lastly, there was a subset of patients who had large doses of radiation and later developed malignancies within the field of radiation [5, 6].
19.2.2.2 Timing of the Radiation Therapy
The timing of radiation therapy plays a role in the formation of second malignancies. Receiving radiation treatment in the first year of life may place the patient at a greater risk of second tumors within the field of radiation than if the radiation is delayed until after 1 year of age. This remains controversial based on what is defined as being within the radiation field. Solely considering those tumors located within the radiation field, there appears to be no affect on age at onset of the second malignancies. However, if the definition is expanded to tumors within the head and neck area including the thyroid, pineal gland, and brain tumors, there appears to be a significant age-related risk. Radiation should therefore be delayed until 1 year of age or avoided altogether if at all possible [5, 7–10].
19.2.2.3 Increased Incidence
Although a relationship exists between prior radiation therapy and the development of a second malignancy, this relationship is neither linear nor definite. One long-term study of heritable retinoblastoma survivors reported an increased cumulative incidence of secondary malignancies (38 %) for those with external beam radiation therapy exposure compared with heritable retinoblastoma survivors who did not receive radiation (21 %) [11]. The increased risk of developing second non-ocular malignancies in patients with heritable retinoblastoma due to radiation exposure [7–9] is not observed in patients with nonheritable retinoblastoma [4, 12–14]. A recent study suggests that proton radiotherapy has a lower rate of radiation-induced second malignancy compared with photon radiotherapy, but longer follow-up is needed to confirm this finding [15].
19.2.2.4 Age of Onset
The onset of non-ocular tumors is variable and increases in incidence with age. Osteosarcomas will usually develop in retinoblastoma survivors during the growth-spurt years, not significantly different from the normal population [12–14]. However, studies suggest that there may be a bimodal distribution between the age of 5–7 years and then a second incidence peak in the early teenage years for retinoblastoma survivors, whereas sporadic osteosarcoma tends to occur in the later teenage years [14].
19.2.2.5 Location
The location of non-ocular tumors is variable and corresponds with the tumor’s cell of origin. Overall, about 70 % of the tumors occur in the head and neck region [4, 9]. However, osteosarcoma, the most common second malignancy, may occur outside this region with a predilection for the long bones of the lower extremities (Fig. 19.2) [4].
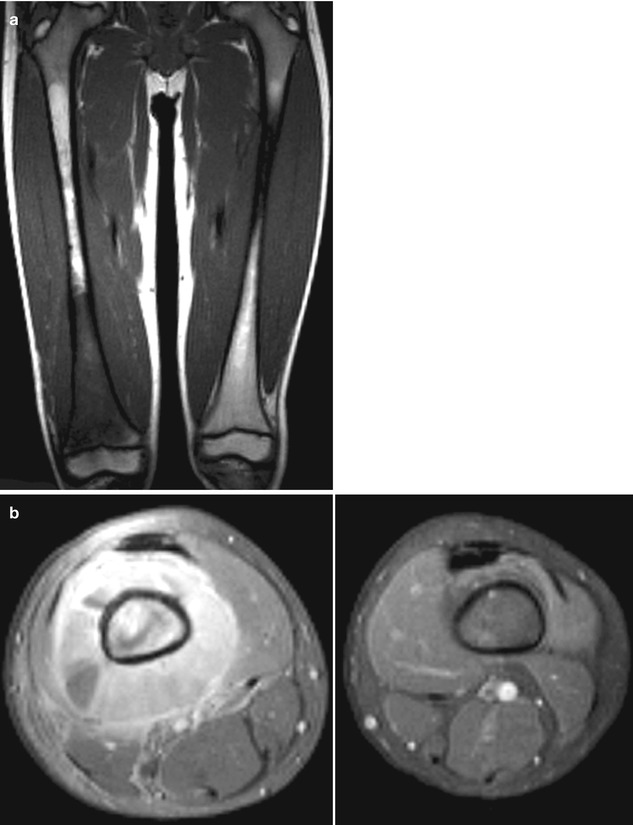
Fig. 19.2
A 7-year-old boy treated for bilateral retinoblastoma during the first year of life who presented with right thigh pain and swelling. A coronal T1-weighted (a) and transverse plane (b) MRI show a mass arising from right distal femur. Biopsy confirmed a primitive neuroectodermal tumor
19.3 Types of Second Malignancies
Two major types of second malignancies are observed in patients with childhood cancers – radiation-associated solid tumors and acute myeloid leukemia and myelodysplastic syndrome related to chemotherapy exposure (alkylating agents and topoisomerase-II inhibitors).
19.3.1 Radiation-Associated Solid Tumors
The most common secondary malignancies in retinoblastoma survivors are solid tumors. Osteosarcomas, both inside and outside the radiation field, make up one-third of the second malignancies; soft tissue sarcomas and melanomas are the next most common.
19.3.2 Alkylating Agent- and Topoisomerase-II Inhibitor-Related Acute Myeloid Leukemia and Myelodysplastic Syndrome
Retinoblastoma is a chemosensitive tumor; chemoreduction combined with intensive focal consolidation therapies is a desirable regimen in lieu of radiation. Etoposide, one of the three main active chemotherapeutic agents, has a known risk of inducing hematopoietic second malignancies, specifically acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) [16]. Long-term follow-up in retinoblastoma has documented reports of AML associated with etoposide use [17]. In an effort to minimize the use of etoposide and thus the small but real risk of a secondary leukemia, etoposide use may be reduced or minimized in multi-agent chemotherapy regimens [17]. Topotecan may be an effective alternative, but continued follow-up will be necessary to adequately evaluate the long-term effects of this regimen [18].
19.4 Incidence
In the United States, mortality associated with retinoblastoma is more commonly related to non-ocular tumors than the primary eye tumor itself. Reports of the cumulative incidence of second cancers in patients with germ-line mutations of the RB1 gene vary, but it is believed to be approximately 1 % per year of life. The 10-year incidence of second malignancies in hereditary retinoblastoma is about 8 % increasing to almost 50 % at 50 years of age (Chap. 14) [13, 14, 19, 20]. Kleinerman et al. compared subsequent cancer risk in 1072 presumed inherited germ-line RB1 mutation retinoblastoma patients (bilaterality and positive family history) with that in 780 sporadic retinoblastoma patients (unilateral and no family history) and found a 37 % increased risk of second cancers in the inherited mutation group [21]. The heritable patients had a cumulative risk of 47 % of developing a new cancer 50 years after diagnosis. Patients in each group showed similar rates of bone and soft tissue cancers, but the hereditary retinoblastoma group had a higher incidence of cutaneous melanoma.
Another study performed by Shinohara et al. evaluated the risk of subsequent malignant neoplasms in survivors of retinoblastoma using the Surveillance, Epidemiology, and End Results (SEER) database [22]. A total of 59 patients were included in the analysis. The cumulative incidence of secondary malignancy at 30 years for patients with unilateral and bilateral retinoblastoma was 1.7 and 28.5 %, respectively (P < 0.001). Patients with bilateral retinoblastoma treated with and without radiotherapy both experienced an increased risk of secondary malignancies. Within the cohort of patients, second malignant neoplasms accounted for 52 % of deaths.
19.5 Clinical Features
A wide variety of neoplasms have been described in retinoblastoma survivors. Not only are these patients at risk for second non-ocular tumors, but there is a lifelong risk for the development of additional third, fourth, and fifth non-ocular cancers [7, 13, 22, 23]. As mentioned, the most common second malignancy is osteosarcoma, which accounts for approximately one-third of the cases [13]. Soft tissue sarcomas and melanomas are second in frequency, accounting for 20–25 % of the cases. Hematopoietic tumors such as non-Hodgkin’s lymphoma and leukemia and sebaceous gland carcinomas of the eyelid have also been reported.
In recent years, it has become apparent that patients with heritable retinoblastoma are also at risk of developing epithelial cancers late in adulthood [8]. Of those, lung cancer appears to be the most common, followed by bladder cancer [4, 9, 24]. This is not surprising, since somatic mutations of the RB1 gene are known to contribute to the development of lung cancer [4, 24, 25]. Finally, an interesting observation is the increased incidence of lipomas in survivors of hereditary retinoblastoma. The incidence of a second neoplasm appears to be higher in those patients with lipomas, suggesting that the presence of lipomas could be a clinical marker of susceptibility to second neoplasms [26].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


