Chapter 62 Neurometabolic disease and the eye
Many neurometabolic diseases present in childhood with ophthalmic manifestations. In some conditions, the characteristic ophthalmic features may lead toward an early diagnosis. In others, the ophthalmic complications present later in the course of the disease but can have significant visual effects for the patient. Findings such as vertical or horizontal gaze palsy, characteristic corneal changes, cherry-red spot, retinopathy, or optic atrophy, particularly in the presence of progressive systemic or neurologic disorders, should alert the ophthalmologist to the possiblity of a neurometabolic disorder.1
Lysosomal disorders
Lysosomal storage disorders (LSDs) arise as a result of defects in lysosomal enzymes, receptor targets, activator proteins, membrane proteins, or transporters causing accumulation of specific substrates within lysosomes (Table 62.1). This leads to impairment of cellular and tissue function.2 Most LSDs have central nervous system (CNS) and systemic defects; untreated, many result in death in infancy or childhood. Whilst each type of LSD is rare, their total incidence is 1 per 7000 to 8000 live births. Many have ocular manifestations present early in the course of the disease (Box 62.1). The pediatric ophthalmologist has an important role in facilitating early diagnosis so appropriate treatment can be started early.
Table 62.1 – Classification of LSD by lysosomal function affected
| Lysosomal function affected | Disorder |
|---|---|
| Metabolism of glycosaminoglycans | Mucopolysaccharidoses |
| Degradation of glycoproteins | Aspartylglucosaminuria, fucosidosis, mannosidosis, Schindler’s disease, sialidosis type 1 |
| Degradation of glycogen | Pompe’s disease |
| Degradation of sphingolipids | Fabry’s disease, Farber’s disease, Gaucher’s disease, GM1 gangliosidoses, GM2 gangliosidoses, Krabbe’s disease, metachromatic leukodystrophy, Niemann-Pick disease types A and B |
| Degradation of polypeptides | Pycnodysostosis |
| Degradation or transport of cholesterol esters or complex lipids | Niemann-Pick type C, Wolman’s disease, cholesterol ester storage disease |
| Multiple deficiencies of lysosomal enzymes | Multiple sulfatase, galactosialidosis, mucolipidosis types I and II |
| Transport and trafficking defects | Cystinosis, mucolipidosis IV, sialic acid storage disorder, chylomicron retention disease with Marinesco-Sjögren’s syndrome, Hermansky-Pudlack syndrome, Chediak-Higashi syndrome, Danon’s disease |
| Unknown defects | Geleophysic dysplasia, Marinesco-Sjögren’s syndrome |
From Wilcox WR. Lysosomal storage disorders: the need for better pediatric recognition and comprehensive care. J Pediatr 2004; 144: S3–S14.
All LSDs have autosomal recessive inheritance except for Danon’s disease, Fabry’s disease and Hunter’s disease (mucopolysaccharidosis type II) which are X-linked recessive. Some disorders are more prevalent in a particular geographic area or in certain populations, such as Gaucher’s, Tay-Sachs, Niemann-Pick type A, and mucolipidosis IV, which are 50−60 times more prevalent in the Ashkenazi Jewish population.2
There is often a wide variation in phenotype, including severity of symptoms, systems affected, and presence of CNS manifestations. In many disorders such as Gaucher’s and Tay-Sachs, there are infantile, juvenile, and adult forms. There are characteristic ocular features in some of the LSDs (see Box 62.1) which should alert the ophthalmologist to their possibility, particularly when seen in a child with suggestive systemic features such as organomegaly, skeletal abnormalities or joint stiffness, coarsening of facial features, or a progressive neurologic or muscular deterioration, or unexplained pain.
Mucopolysaccharidoses
The mucopolysaccharidoses (MPSs) are a heterogeneous group of disorders resulting from accumulation of glycosaminoglycans within ocular and systemic tissues. There is a wide spectrum of phenotypes with a range of skeletal, cardiac, respiratory, gastrointestinal, and neurologic manifestations. The MPSs have been classified according to phenotype, and result from mutations in different lysosomal enzymes (Table 62.2). They are all inherited in an autosomal recessive manner apart from MPS II (Hunter’s) which is X-linked.
Early hematopoietic stem cell transplantation and ERT have improved the prognosis for many MPS patients. Visual impairment is common, and can be due to corneal opacification, optic neuropathy, retinopathy, or cortical visual impairment.3
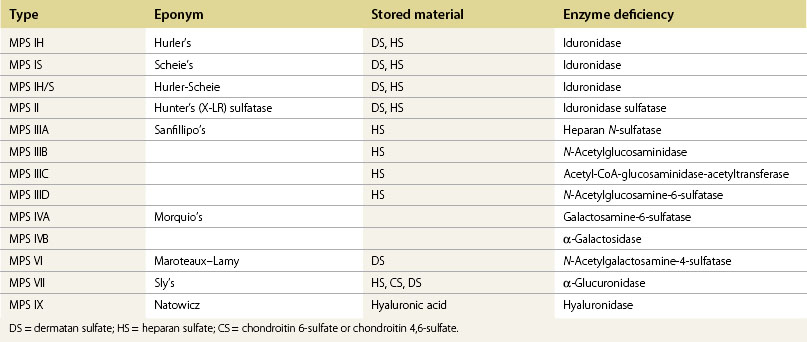
Corneal clouding in MPS
Corneal clouding is a characteristic feature of several of the MPS disorders (MPS I Hurler’s, Hurler/Scheie and Scheie’s, MPS IV Morquio’s, MPS VI Maroteaux-Lamy, and MPS VII Sly’s), and may help facilitate the diagnosis when present at an early stage. Deposition of glycosaminoglycans (GAGs) within the corneal stroma leads to progressive diffuse corneal opacification, described as having a “ground-glass” appearance (Fig. 62.1). Corneal opacification is milder in the less severely effected MPS I phenotype Scheie’s, and is not a feature of MPS III Sanfillipo. Patients with MPS may have coarsening of facial features with pseudoexophthalmos (Figs 62.2 and 62.3), which can result in corneal exposure and subsequent vascularization.
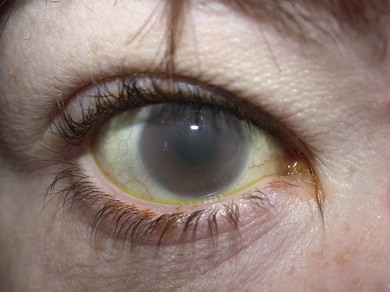
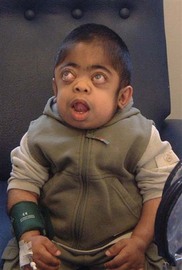
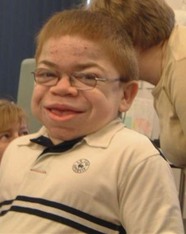
A patient with mild corneal clouding may be asymptomatic, but photophobia and reduced vision occur as the opacification worsens. GAG deposition can increase corneal thickness4,5 and affect corneal hysteresis,6 thus affecting the accuracy of intraocular pressure measurements. Corneal transplantation (penetrating keratoplasty or deep lamellar keratoplasty) may result in improvement in vision when severe corneal opacification causes visual loss7,8 with no reopacification of the graft in up to 11 years follow-up.7 However, an assessment of potential benefits of corneal transplantation must consider the presence of coexistent retinopathy, optic nerve dysfunction, and cortical visual impairment, as well as the significant anesthetic risks for a patient with MPS.
Hypermetropia and strabismus in MPS
The majority of patients with MPS are hypermetropic due to changes in corneal refraction and reduced axial length.3,9 Strabismus and amblyopia are common.3
Retinopathy in MPS
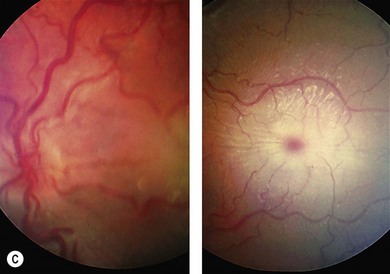
Progressive retinopathy occurs in MPS I, II, III but is not usually a feature of MPS VI.10 The patient may experience night-blindness and problems with peripheral vision, but this may go unnoticed due to the visual loss associated with corneal opacities. Signs of retinopathy include retinal pigment epithelial (RPE) atrophy and pigment clumping (Fig. 62.4), arteriolar narrowing, optic disk pallor, and macula changes. Serial electroretinography (ERG) may show initial deterioration of rod mediated responses, followed by involvement of cones. Choroidal folds have occurred in MPS II associated with a ring scotoma; optical coherence tomography demonstrated extrafoveal photoreceptor loss and cystoid spaces within retinal layers.11
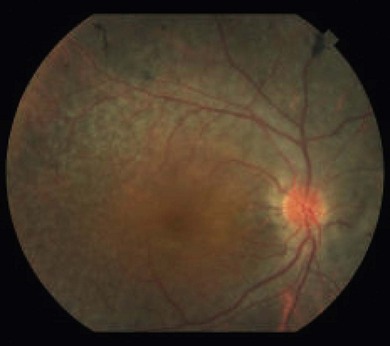
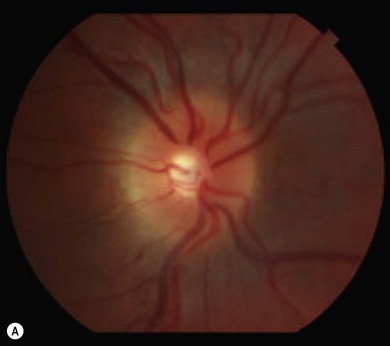
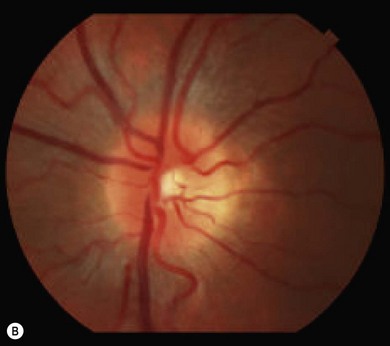
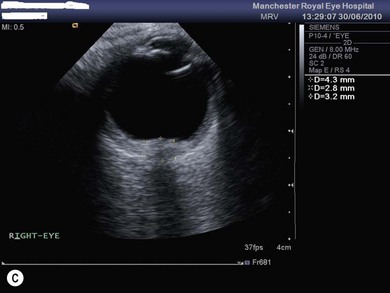
Optic disk swelling and atrophy
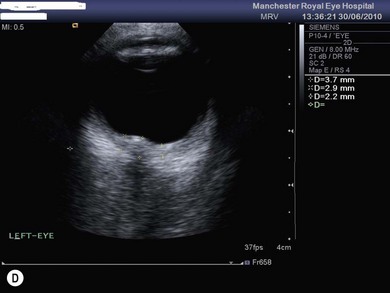
The optic nerve often has a “full” appearance in patients with MPS (Fig. 62.5A,B). GAG deposition occurs in the ganglion cells and within surrounding sclera causing an enlargement of the nerve and thickening of the sclera12 (Fig. 62.5C,D). The consequent nerve compression can lead to optic atrophy. In addition, patients with MPS may develop raised intracranial pressure and the resultant optic nerve compromise may lead to profound visual loss. Visual evoked potential may be useful in monitoring optic nerve function and in assessment prior to consideration of corneal transplant.
Glaucoma
Glaucoma may arise in MPS due to accumulations of GAGs within the anterior segment resulting in a narrow angle, and within the trabecular cells leading to open angle glaucoma as a result of outflow obstruction. It has been described in MPS I, II, and VI.10 Difficulties in diagnosis and assessment of glaucoma arise in patients with MPS due to inaccuracy of intraocular pressure measurements, corneal clouding hampering assessment of the angle and disk, and the presence of coexistent disk pathology.
Systemic manifestations in MPS
A patient with MPS may present in infancy with recurrent ear infections, inguinal or umbilical hernias, or skeletal changes such as kyphoscoliosis (Fig. 62.6). Other systemic manifestations include obstructive sleep apnea, deposits on the cardiac valves and infiltration of the cardiac muscle, spinal cord compression, and pain and stiffness from skeletal dysplasia. Developmental delay and intellectual impairment occur in MPS I; behavioral problems occur early in MPS III, as a result of CNS deposition of GAG. Management is multidisciplinary, coordinated by a pediatrician with input from cardiology, anesthesia, orthopedics, ENT, neurosurgery, physiotherapy, audiology, speech therapy, and ophthalmology. Children with a diagnosis of MPS should have regular assessments by a pediatric ophthalmologist.

Prognosis and treatment of MPS
The prognosis in MPS is variable depending on the phenotype; some children die before their second decade and others survive into their fifth or sixth decade. Early hematopoietic stem cell transplantation using either matched bone marrow or umbilical cord blood cells can improve the systemic prognosis of patients with MPS I and VI. ERT for MPS I (laronidase), MPS II (elaprase), and MPS VI (galsulfase) improves systemic manifestations and can be used prior to early BMT. Ophthalmic manifestations of MPS VI may be stabilized by ERT.13
Neuronal ceroid lipofuscinosis
The neuronal ceroid lipofuscinoses (NCLs) are autosomal recessive conditions characterized by progressive neurologic deterioration in children or young adults, epilepsy, and visual deterioration due to retinopathy. All the NCLs are associated with accumulation of an autofluorescent material, ceroid lipofuscin, and degeneration of neuronal cells.14 There are at least 10 genetically distinct NCLs (Table 62.3). The term Batten’s disease refers to the juvenile-onset form of NCL and occurs most commonly as a result of mutations in CLN3, which maps to chromosome 16p21.
Diagnosis of NCLs can be made by demonstration of specific enzyme deficiency in leukocytes, fibroblasts or blood, such as palmitoyl protein thioesterase, which can be used for diagnosis of NCL1.14 In a school child with retinopathy, CLN3 diagnosis may be confirmed by finding vacuolated lymphocytes in the peripheral blood smears. The more rare NCL variants can be demonstrated by electron microscopic examination of skin biopsy material or isolated blood lymphocytes before proceeding to molecular genetic testing.14
Juvenile neuronal ceroid lipofuscinosis (CLN3, Batten’s disease)
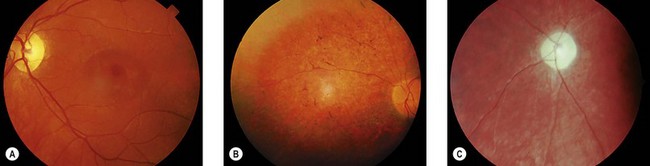
Children with juvenile NCL present with rapid deterioration of vision between 4 and 10 years. Other manifestations such as behavioral problems and cognitive decline and motor deterioration manifest before or after the onset of visual loss.15–17 There is an increased prevalence in North European populations.15 Night blindness and photophobia may be present in addition to visual loss. Misdiagnosis as macula or retinal dystrophy or even functional visual loss may occcur in the early stages.16 At presentation, the fundus may be normal or show pigmentary maculopathy, atrophic changes, or bull’s-eye maculopathy. Later there is a widespread retinal degeneration with pigment clumping (Fig. 62.7) and sparse bone-spicule pigmentation in the periphery, the disk becomes atrophic and the arterioles thinned (Fig. 62.7C), and the retina becomes avascular. Eccentric viewing or “overlooking” is where children hold their eyes in a raised position whilst attempting to fixate on a target, presumed due to the relative preservation of the superior peripheral retina.16,17 The children are usually blind within 3 years. Although mental deterioration and behavioral disturbances occur early, often predating the visual deterioration, they are often quite subtle. The disease follows a slow downward path with the onset of fits between 7 and 16 years of age, dementia in the teens, and death sometimes in the second or third decade.
Fluorescein angiography shows diffuse RPE atrophy with stippled hyperfluorescence.18 ERG may show a photopic and scotopic electronegative waveform, typically in early cases with a markedly reduced b : a ratio in the single flash photopic ERG, consistent with inner retinal dysfunction.15,19 ERG changes may prompt further investigations to be carrried out at an early stage. Visual field testing may show field constriction.
Glycoprotein disorders
α-Mannosidosis (α-mannosidase deficiency)
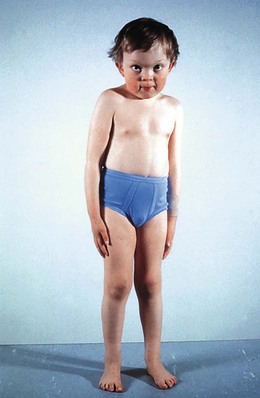
α-Mannosidosis is associated with mild facial coarsening (Fig. 62.8), skeletal abnormalities, a variable degree of learning difficulty, and deafness. Ophthalmologic features include cataracts and strabismus.20 The lens opacities are posterior cortical with multiple discrete clear round vacuoles lying at different depths in the lens, best seen by slit-lamp retroillumination.21
Fucosidosis
Fucosidosis is a progressive neurodegenerative disease with seizures, mild coarsening of the facies, skeletal dysplasia, and angiokeratoma. Affected children have conjunctival and retinal vascular tortuosity. A bull’s-eye maculopathy and central “lobulated” corneal opacities may occur.22
Sialidosis (mucolipidosis type I)
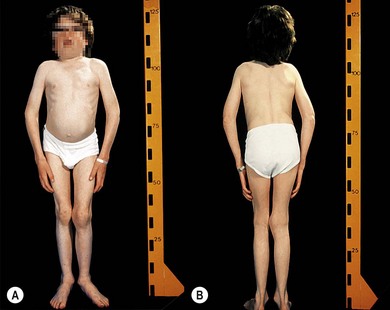
Type 1 sialidosis “cherry-red spot myoclonus syndrome” presents in late childhood with visual loss associated with a cherry-red spot (Fig. 62.9), myoclonic epilepsy, and ataxia. Type 2 sialidosis has an earlier onset with coarse facies, developmental delay, hepatomegaly, and dysostosis multiplex (Fig. 62.10).
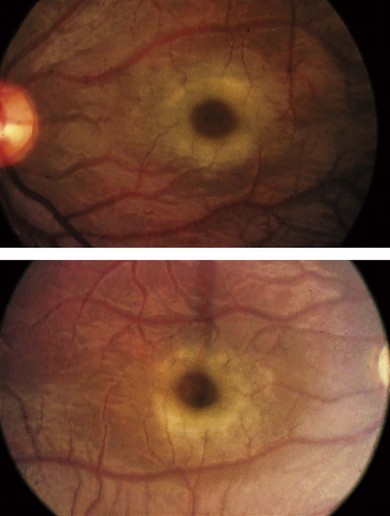
In addition to cherry-red spots, patients with sialidoses may have punctate lens opacities, optic atrophy, corneal clouding, and visual field defects.23–25
Sialic acid storage diseases
These result from a block in lysosomal efflux of sialic acid as a result of mutations in SLC17A5, encoding the lysosomal protein sialin. They range in severity from infantile sialic acid storage disease (ISSD) with coarse facial features and fair complexion, severe developmental delay, hepatosplenomegaly and cardiomegaly, and death in early chidlhood, to Salla’s disease with slowly progressive mental retardation, ataxia, spasticity, and epilepsy. In ISSD, children may have albinoid fundi.26 In Salla’s disease strabismus, nystagmus, and optic atrophy may occur.
Mucolipidoses
ML II or “I cell” disease presents at an early age with neurologic degeneration, joint stiffening, facial coarsening, and kyphoscoliosis (Fig. 62.11). Death occurs in childhood. There is retinal degeneration and corneal clouding.

ML III is milder, with a slowly progressive course and survival to adulthood. Ophthalmic features include corneal clouding, hypermetropic astigmatism, optic disk swelling, retinal vascular tortuosity, and maculopathy.27 The ERG is normal.
ML IV is an autosomal recessive disorder resulting from mutations in the MCOLN1 gene, which encodes mucolipin-1. It is seen mainly in Ashkenazi Jews. Clinical features include progressive mental retardation and hypotonia. ML IV patients have corneal eipthelial haze, retinopathy with optic nerve pallor, vascular attenuation, and RPE change with electronegative ERG.28,29 Other ophthalmic features include strabismus, corneal erosion and episodic pain, cataract, and ptosis.28,30 Mild phenotypes have been described with only ocular manifestations of corneal clouding and retinopathy.31,32
Sphingolipidoses
GM2 gangliosidosis
• Tay-Sachs disease: hexosaminidase A deficiency, AR, most common in the Ashkenazi Jewish population.
A cherry-red spot is apparent at an early stage, due to GM2 ganglioside accumulation in the retinal ganglion cells. As the ganglion cells die the cherry-red spot fades and optic atrophy becomes apparent.33 The ERG is normal, but the visual evoked potential extinguished.34 There is no treatment.
Both disorders have a later-onset variety; ataxia and dementia begin in the second year of life and the disease follows a more attenuated form. Late onset Tay-Sachs disease presents in an adult and is associated with abnormalites of saccades: saccadic hypometria, transient decelerations, and premature terminations.35 A cherry-red spot is less common in the late onset types (Table 62.4).
Table 62.4 – Cherry-red spot in neurometabolic disorders1
| Disease | Disease frequency of cherry-red spot |
|---|---|
| Niemann-Pick type A | Occasional |
| Niemann-Pick type B | Occasional |
| GM2 gangliosidosis (Tay-Sachs and Sandhoff’s) | Frequent |
| GM1 gangliosidosis, infantile | Occasional |
| Galactosialidosis | Frequent |
| Farber’s lipogranulomatosis | Occasional |
| Sialidosis type 1 | All |
| Sialidosis type 2 | Frequent |
| Metachromatic leukodystrophy | Occasional |
GM1 gangliosidosis
Infantile GM1 gangliosidosis causes hypotonia from birth. By 6 months, developmental delay and a coarse appearance with puffy skin, maxillary hyperplasia, hypertrophied gums, and macroglossia develop. Affected children have dysostosis multiplex. Ocular motor apraxia (saccadic initiation failure),36 a cherry-red spot, and corneal clouding may occur. Rapid neurologic deterioration is usual with seizures and swallowing difficulties and death by 2 years. There is no treatment. Juvenile and adult forms occur in which there is neurologic deterioration but no physical changes.



