CHAPTER 60 Neurologic Disorders of the Larynx
Laryngologic Manifestations of Neurologic Diseases
Cortical lesions may result from strokes, tumors, or trauma and may impair memory as well as the planning and execution of actions. Because of the diffuse and bilateral representation of laryngeal structures in the cortex, cortical lesions, such as those caused by tumors or strokes, do not produce classic flaccid or spastic paralysis. Inappropriate vocal fold adduction with inspiration may have the symptom inspiratory stridor.1,2 Extrapyramidal system defects are characterized by abnormal motor control, which may manifest as inappropriate or excessive muscle tension, tremor, and involuntary spasmodic muscle contractions. Vocally, it translates into strain, arrests, pitch breaks, and pitch instability. The dysfunction may be focal, regional, or generalized. In addition to problems caused by tumors or trauma, the extrapyramidal system is disrupted by conditions of uncertain etiology such as Parkinson’s disease, tremor, and dystonia.3 Cerebellar lesions impair coordination of motor activities. Patients’ problems are generalized rather than focal. “Scanning speech” is regarded as characteristic of cerebellar involvement. Diagnosis is based on the presence of attendant physical signs, such as intention tremors, dysdiadochokinesia, dysmetria, ataxia, and nystagmus.4 Brainstem lesions result in flaccid paralysis. Because the cranial motor nuclei are densely packed within the brainstem, lesions at this level affect multiple outputs. Strokes and tumors of the brainstem produce severe dysfunction related to paralysis of the larynx, pharynx, or tongue and are associated with sensory deficits.2 The site of lesion is best identified from the type of motor disruption, because observable clinical signs are predominantly disruptions of motor acts (Table 60-1).5,6 Diffuse central nervous system (CNS) lesions result from specific neurologic disorders, such as multiple sclerosis and amyotrophic lateral sclerosis (ALS), and have a myriad of signs and symptoms. Patients with movement disorders have a paucity of movement (akinesia or bradykinesia), excessive or hyperfunctional movement (hyperkinesia), or a combination of the two. The hyperkinetic motor programming errors can produce spasms, tremors, jerks, or tics as well as symptoms related to the body part involved. For those with laryngeal manifestations, a multidisciplinary approach that includes the involvement of an otolaryngologist, a neurologist, and a speech pathologist is key to successful diagnosis and management of hyperfunctional disorders of the larynx.7,8
Table 60-1 Signs Indicating Site of Lesion in Neurologic Disorders of the Larynx
| Site of Lesion | Signs |
|---|---|
| Cortex | |
| Extrapyramidal system | |
| Cerebellum | |
| Brainstem |
Hyperfunctional Disorders
Dystonia
Dystonia is a syndrome dominated by sustained muscle contractions of skeletal muscles that frequently causes twisting and repetitive movements or abnormal postures that may be sustained or intermittent. Because the condition is rare and the movements and resulting postures are often unusual, dystonia is among the most commonly misdiagnosed neurologic conditions.9 The prevalence is unknown, but there are an estimated 50,000 to 100,000 cases of idiopathic dystonia in the United States. Classification is important because it can inform prognosis and the approach to management. The classification scheme is outlined in Box 60-1.
Dystonia can begin at nearly any age. Presenting signs have occurred as early as 9 months and as late as 85 years. In general, onset has a bimodal distribution, with peaks at ages 8 and 42 years. Early-onset dystonia begins to manifest before age 26 years, whereas in the late-onset form, the signs appear at older ages. Patients are also categorized according to symptom distribution. Focal dystonia involves one small group of muscles in one body part, segmental disease involves a contiguous group of muscles, and generalized dystonia is widespread. The more common examples of focal dystonia are listed in Box 60-2.7
History, physical examination, and laboratory studies may not identify the cause of a patient’s dystonic symptoms (idiopathic dystonia). Therefore, there should be a normal perinatal and early developmental history; no history of neurologic illness or exposure to drugs known to cause acquired dystonia (e.g., phenothiazines); normal intellectual, pyramidal, cerebellar, and sensory examination findings; and normal diagnostic study results. The clinical phenomenology is often a clue to the cause. Primary dystonia is typically action induced and normal at rest, but secondary dystonia frequently results in fixed dystonic postures. The presence of extensive dystonia limited to one side of the body (hemidystonia) also suggests a secondary cause.7,9
Because up to 16% of patients with dystonia and primary laryngeal involvement experience spread of the disease to another body part, patients should be advised of this potential and should be reexamined on a regular basis for signs of other dystonic involvement. Approximately 10% of patients with primary laryngeal dystonias have a family history of dystonia.10
In most cases of childhood-onset idiopathic dystonia, family studies show an autosomal dominant inheritance with reduced penetrance. A marker for some cases of childhood-onset dystonia has been found on chromosome 9.11 There are heterogeneous genetic patterns among patients with idiopathic dystonic symptomatology, including a linkage to the X chromosome and parkinsonism,12–14 a dopamine-responsive form.15
Family and linkage studies have identified several subtypes with different genetic bases, including the autosomal dominant dopamine-responsive dystonia already mentioned,15 X-linked Filipino torsion dystonia,12–14 and an autosomal dominant (non–dopamine-responsive) idiopathic torsion dystonia related to the DTY1 gene mapped to chromosome 9q34. Both dopamine-responsive dystonia and X-linked torsion dystonia are rare forms of idiopathic torsion dystonia associated with parkinsonism.6,11,16,17
Clinically, spasmodic dysphonia is an idiopathic focal dystonia of the larynx.17a Although the disease was initially described by Traube18 in 1871, Fraenkel19 and Gowers20 later recognized the relationship with other dystonias, such as writer’s cramp. The vast majority of cases of laryngeal dystonia cause spasmodic adduction of the vocal folds during speech, which results in a strained and strangled voice. Fewer patients have abductor dysphonia, with intermittent or sustained opening of the larynx during speech21 that leads to breathy voice breaks or a whispering voice. Some patients display a combination of adductor and abductor signs and have been classified as having “mixed laryngeal dystonia.”7,22 Patients with spasmodic dysphonia have reported that symptoms momentarily improve when he or she pinches the nares, presses the hand against the back of the head, presses the hand into the abdomen, pulls on an ear, or touches the clavicular notch (personal experience). Many patients observe that they speak better after a yawn or sneeze or when they sing or yell; these sensory tricks are also common for patients with other craniocervical dystonias.9,23 Systemic pharmacotherapy provides little relief of symptoms. Dedo and Izdebski24 described dramatic relief of symptoms with sectioning of the recurrent laryngeal nerve. The initial favorable reports were temporized by a review of 33 patients by Aronson and De Santo3,25 that addressed surgical management. Three years later, only 36% of patients had some persistent improvement, and only 3% achieved a persistent normal voice. At present, the symptoms are most effectively managed with use of an individualized regimen of chemodenervation with botulinum toxin.
Another rare form of laryngeal dystonia is adductor breathing dystonia, in which patients adduct their vocal cords while inspiring. The adduction causes stridor and dyspnea, but these patients do not become hypoxic and do not need tracheostomy. Localized injection of botulinum toxin into involved muscles is an effective way to diminish the symptoms of patients with dystonia, particularly those with adductor spasmodic dysphonia.26–30 Patients with more generalized dystonia that also involves the larynx have vocal dysfunction, which is clinically indistinguishable from idiopathic spasmodic dysphonia. Meige’s syndrome, a regional dystonia of the head and neck, may be evident in those with blepharospasm, oromandibular dystonia, torticollis, or spasmodic dysphonia.
Botulinum Toxin Therapy
The bacterium Clostridium botulinum produces eight immunologically distinct toxins that are potent neuroparalytic agents: A, B, C1, C2, D, E, F, and G.31,32–36 Botulinum toxin exerts its effect at the neuromuscular junction by inhibiting the release of acetylcholine, causing a flaccid paralysis.4,37–39 Botulinum toxin type A (Botox; Allergan, Inc., Irvine, CA) is most commonly used, and type B (Myobloc; Elan Pharmaceuticals, Inc, San Francisco) is also available for clinical application.
Although botulinum toxin has been used therapeutically in humans since the mid-1970s without evidence of a direct effect on uninjected muscles, the later consequences of long-term injections are unknown. Weakness and routine electromyography (EMG) changes in muscles distal to the site of injection have not been reported. However, there are detectable abnormalities on single-fiber EMG.40 It is not known how long these abnormalities persist or whether they have any clinical significance. There is a paucity of data regarding use of botulinum toxin during pregnancy. Currently, injection should be avoided in pregnant or lactating patients. Caution is warranted for the management of patients with conditions such as myasthenia gravis, Eaton-Lambert syndrome, and motor neuron disease, particularly when large doses are required (e.g., in the management of cervical dystonia). However, the amount of toxin entering the circulation after injection is thought to be minute, and this theoretic concern should be balanced against the severity of the hyperkinetic symptoms.40
In patients with adductor spasmodic dysphonia, botulinum toxin injection has been demonstrated to improve speaking to 60% to 100% of normal function,41 with a mean of 90%; the duration of effect was between 3 and 4 months. Adverse effects include a mild breathy dysphonia for less than 2 weeks (45%), mild choking on fluids for the first several days (22%), hyperventilation and dizziness when trying to speak while hypophonic, a sore throat or coughing up blood-tinged sputum, and itching (without rash).42–4581 In patients with abductor laryngeal dystonia, injection of botulinum toxin into the posterior cricoarytenoid muscle produces marked improvement, with a return to mean maximal functional performance of 70% of normal. Adverse effects include mild dysphagia without aspiration and mild stridor on exertion.
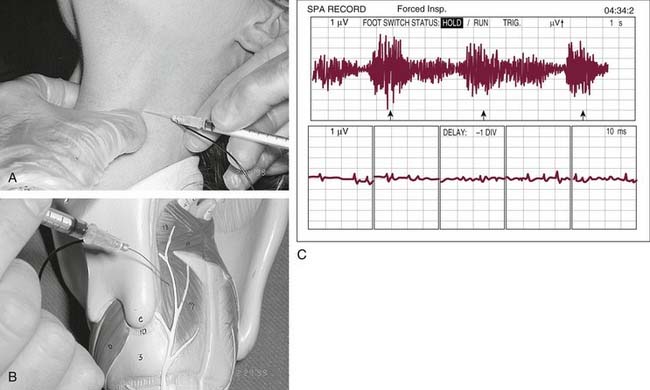
The effective treatment dose is variable for each patient and for each muscle injected; therefore, injections are individualized.46 The dose range for adductor spasmodic dysphonia is 0.05 to 20 units of Botox, with an average dose of less than 1 unit per vocal fold.24,29,30,47,48 In general, a starting dose of 1.0 units in 0.1 mL of saline is used for bilateral thyroarytenoid injections. Subsequent doses are varied according to clinical response and adverse effects. A trend toward lower doses has been observed and may be useful in reducing adverse effects.49 Injections are given by means of a tuberculin syringe with a 27-gauge monopolar polytef-coated hollow EMG recording needle. Adductor laryngeal injections are performed percutaneously through the cricothyroid membrane and into the thyroarytenoid-vocalis muscle complex, with use of EMG guidance for optimum placement (Fig. 60-1). Injections for abductor spasmodic dysphonia are administered to the posterior cricoarytenoid muscle. The physician reaches muscle by manually rotating the larynx, placing the EMG needle behind the posterior edge of the thyroid lamina, and advancing the needle to the cricoid cartilage to arrive at the posterior cricoarytenoid muscle. Alternatively, a transcricoid injection can be made. When the patient is instructed to sniff, which maximally uses the posterior cricoarytenoid muscle, there is a burst of activity on the EMG, and the toxin is administered.42,43,47,50
After an injection, the patient typically reports improvement in voice within 24 hours followed by a breathy, hypophonic period lasting 1 to 2 weeks (45%), occasionally causing hyperventilation and dizziness when trying to speak. Mild choking on fluids for the first several days (22%) is also common; in addition, sore throat or coughing up blood-tinged sputum may occur in the first 1 to 2 days after injection.42–44
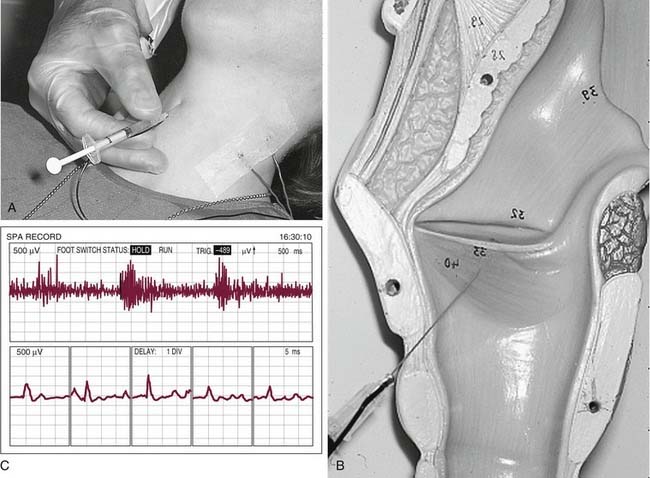
Patients with abductor spasmodic dystonia are also managed with an EMG-guided percutaneous injection. The larynx is manually rotated away from the side of intended injection, and the hollow EMG needle with syringe is placed posterior to the posterior edge of the thyroid lamina (Fig. 60-2).