CHAPTER 64 Laryngeal and Tracheal Manifestations of Systemic Disease
Wegener’s Granulomatosis
Wegener’s granulomatosis is a multisystem inflammatory disease characterized by necrotizing granulomatosis and necrotizing vasculitis of small arteries, arterioles, capillaries, and venules.1,2 It primarily involves the upper and lower respiratory tracts, including the larynx and trachea, and kidneys.1 Other areas of involvement are the oral cavity, skin, joints (polyarteritis), and orbit.1 In the airway, the subglottis and trachea are more commonly affected (Fig. 64-1).
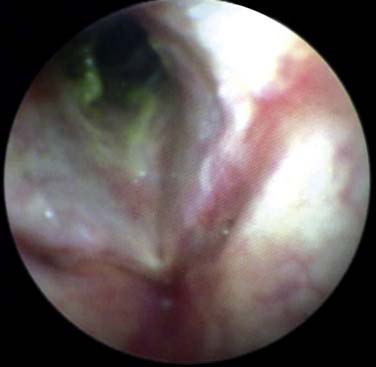
Airway involvement usually occurs in the presence of systemic disease but may be the only sign of the illness. Specifically, 10% to 20% of patients with Wegener’s granulomatosis present with subglottic involvement and subsequent stenosis.1,3,4 As a result, patients may be incorrectly diagnosed as having asthma when stridor is mistaken for wheezing. Glottic involvement manifests as voice changes. Most patients with Wegener’s granulomatosis have a positive ANCA (antineutrophil cytoplasm antibody) test result, but up to 20% of patients who have the disease with respiratory involvement have negative ANCA test results.2
Common symptoms of Wegener’s granulomatosis of the airway include hoarseness, cough, hemoptysis, dyspnea, stridor, and wheezing. Progressive respiratory difficulty with stridor can be the first signal of airway involvement. Pulmonary function testing with flow-volume loops can be helpful in distinguishing a fixed extrathoracic lesion (i.e., subglottic stenosis) from parenchymal disease.2 The typical pattern of flattening of both inspiratory and expiratory phases is seen in fixed lesions.2
Management of the disease is complex and usually requires a multidisciplinary approach. Systemic manifestations are treated with systemic corticosteroids and other immunomodulating agents, depending on disease severity. First-line treatment is with steroids plus cyclophosphamide.2,4 Once remission has been achieved, methotrexate or azathioprine is used.2 Airway disease in Wegener’s granulomatosis presents a complex challenge because it frequently behaves independently of the systemic disease. Treatment should be tailored to the presenting symptoms and level of distress.
In the presence of acute airway obstruction, tracheostomy placement can be lifesaving. With current endoscopic techniques, however, many patients can be managed without tracheostomy.3–5 Although techniques differ, dilation of the stenosis with injection of intralesional steroids is the mainstay of treatment. There are reports of the use of topical mitomycin C after dilation.6 Depending on the severity of narrowing or the level of the stenosis, dilation can be achieved with rigid dilators or balloon dilation.2–4 The carbon dioxide laser has also been used to treat subglottic stenosis.6 In patients with difficult stenoses, stents or tracheostomy can be used. Finally, resection and reanastomosis have been used to remove the stenotic segment, but this step is usually reserved for mature stenoses and inactive disease.
Relapsing Polychondritis
Relapsing polychondritis is a rare inflammatory disease of unknown etiology that causes episodic inflammation of cartilaginous structures throughout the body.7 Commonly affected areas are the ears, nose, eyes, larynx, bronchi, costal cartilages, and articular joints. Males and females are affected equally, with an annual incidence of 3.5 per million.8 All ages can be affected, but peak onset is somewhere in the fourth to fifth decade. Only about 14% of patients present with airway symptoms, but 50% to 55% of patients eventually have them.7 Diagnosis is based on the presence of multiple criteria, first described by McAdam and colleagues7 and later modified by Damiani and Levine.9 The most common finding is bilateral auricle chondritis. Respiratory involvement is less common but more damaging. In 25% to 35% of patients with relapsing polychondritis there is a preceding autoimmune process.8,10
Involvement of the respiratory tract can have serious consequences. Typically patients present with hoarseness, cough, choking, wheezing, dyspnea on exertion, stridor, or tenderness of the laryngeal framework.7–9 Mortality rates are reported anywhere from 10% to 50% when the laryngotracheobronchial tree is involved.8 The airway can be involved at various levels, making treatment difficult. Upper airway obstruction can occur with laryngeal involvement. Collapse of the trachea or bronchi happens when the more distal airway is affected. Respiratory distress may occur by either of two mechanisms, collapse of the central airway from destruction and fibrosis of the laryngeal and tracheal cartilages or peripheral airway narrowing from inflammation and cicatricial fibrosis.11 Patients may ultimately require tracheostomy for airway stabilization.
Imaging can be helpful in the diagnosis of relapsing polychondritis. Plain films can demonstrate the nonerosive arthropathy seen in the disease.9 Airway fluoroscopy has been implemented to evaluate for central airway collapse during expiration.11 Magnetic resonance imaging and computed tomography are complementary in the diagnosis of relapsing polychondritis. Typical computed tomography findings include subglottic stenosis, tracheobronchial luminal narrowing, dense calcification and thickening of tracheal cartilages, peripheral bronchial narrowing, and bronchiectasis11; the most common finding is increased attenuation and smooth thickening of the airway walls.11 Magnetic resonance imaging is very sensitive for visualization of soft tissue inflammation. Such inflammation tends to be hyperintense on T2-weighted images, and to enhance with gadolinium on T1-weighted images.8
Histologic examination shows chondritis with a mixed inflammatory cell infiltrate composed of polymorphonuclear leukocytes, lymphocytes, plasma cells, and eosinophils, depending on the stage at which the lesion is sampled.7,9 The primary change is the loss of matrix mucopolysaccharides followed by a secondary perichondrial inflammatory reaction.12
In addition to surgical measures, relapsing polychondritis is managed with high-dose corticosteroids. Maintenance therapy consists of methotrexate and low-dose corticosteroids. Azathioprine, cyclophosphamide, cyclosporine, and dapsone have been tried in refractory cases.8 There have also been case reports of the use of infliximab, which blocks tumor necrosis factor α, to treat refractory cases.13
Sarcoidosis
Sarcoidosis is a chronic granulomatous disease of unknown etiology. It is characterized by noncaseating granuloma formation with clusters of tubercles in the same stage of development.14,15 It can affect any organ system within the body.14 Nine percent of all patients with sarcoidosis have head and neck manifestations.14 The first histopathologic diagnosis of laryngeal involvement was confirmed by Poe in 1940, despite its earlier description in the literature.14,15 The estimated incidence of laryngeal sarcoidosis is between 1% and 5%.14 Although rare, laryngeal sarcoidosis can be the only manifestation of the disease.
Symptoms are generally mild despite extensive tissue involvement. The progression of the disease is slow and is characterized by many relapses and remissions.15 With respect to laryngeal involvement, symptoms typically include hoarseness, dyspnea, stridor, dysphagia, and cough.14,16,17 However, patients can be asymptomatic. Rarely, airway obstruction may be the presenting symptom.14
The supraglottic larynx is affected most often, followed by the subglottis. The vocal folds are frequently spared. The epiglottis is the most common subsite, followed by the arytenoids, aryepiglottic folds, and false vocal folds.14 When airway involvement is suspected, laryngoscopy is a key step in the diagnosis.
The classic appearance of the larynx affected by sarcoidosis is described as “turban-like thickening” (Fig. 64-2).15 This thickening is due to the pale, pink, diffuse induration and swelling of the supraglottis. Flexible fiberoptic examination may reveal the presence of edema and erythema, punctate nodules, mass lesions, and ulcerations.14 Biopsy of affected areas shows lymphocytic infiltration and noncaseating epithelioid granulomas.14 The pathologic findings can be confused with those in other granulomatous diseases, and additional studies and cultures are recommended.14
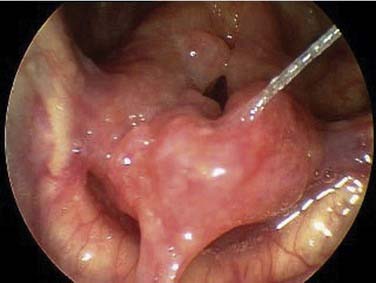
Treatment of the laryngeal manifestations of sarcoidosis can be achieved with systemic corticosteroids. For lesions that are small and well circumscribed, some writers have performed intralesional injection of corticosteroids.14,17 In cases in which there is airway obstruction, excision with the carbon dioxide laser or Microdebrider can be performed to prevent the need for a tracheostomy. It is preferable to treat lesions when they are nonobstructing so as to avoid the need for a tracheostomy.17 Due to the risk of laryngeal involvement, it is recommended that patients with sarcoidosis who begin to have airway symptoms be evaluated by an otolaryngologist.15
Amyloidosis
Amyloidosis is an idiopathic disorder characterized by the extracellular deposition of insoluble fibrillar proteins. Interestingly, the origins of these fibrillar proteins are their normally soluble variants. These deposits were initially described by Rokitansky in 1842.18 The first laryngeal case was described by Borow in 1873.18 Since then it has rarely been described as affecting the larynx, accounting for as little as 0.2% of benign tumors of the larynx.19
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


