CHAPTER 118 Neoplasms of the Neck
Radiologic Studies
Similar to CT, MRI should be performed with contrast enhancement using gadolinium agents. Fat suppression is helpful for postcontrast and T2-weighted sequences; however, nonenhanced T1-weighted images without fat suppression should always be obtained because they frequently provide the best delineation of the normal anatomic structures and the extent of pathologic processes.1,2 Positron emission tomography (PET) with fluorine-18 fluorodeoxyglucose (FDG), primarily PET-CT scanning, has evolved as a well-established modality in the evaluation of the primary site, regional nodal disease, and the presence of distant tumor for staging information, particularly for squamous cell carcinoma and the detection of synchronous primary tumors such as lung and esophagus. PET-CT scans may allow for precise localization of the lesion and may decrease the number of false-positive and false-negative findings.2,3 A controversial use of PET/CT imaging is in the clinical decision making of neck treatment following definitive chemoradiation therapy for advanced head and neck cancer. Several studies have addressed the use of PET scanning in the postirradiated neck. Porceddu and colleagues have shown that in patients with N2 and N3 neck disease a negative PET scan at 8 weeks allowed the observation of those patients with residual neck mass without compromising regional control. Of 27 patients observed by Porceddu with negative PET scans there were no failures in the neck. Yao and associates obtained similar results in a similar patient population. Optimal timing for post-therapy PET scanning appears to be at least 8 weeks, because scanning at 8 to 12 weeks significantly improved the negative predictive value over those who scanned earlier.4–6 On the basis of these studies it may be a reasonable strategy to obtain a PET-CT scan 8 to 12 weeks following therapy to evaluate for residual disease and performing neck dissection on those with positive scans while observing those with negative scans whether a neck mass is present or not. It should be mentioned that the spatial resolution of PET (and PET-CT) allows for detection of lesions that are at least 8 mm in size.
Ultrasound imaging has become a valuable tool in the head and neck surgeons’ diagnostic repertoire. It has the ability to allow the addition of imaging to the in-office examination of the neck as an adjunct to the physical examination. In addition, for the evaluation of the N0 neck it allows not only the characterization of lymph nodes in the neck but allows guided FNA biopsy of nodes of interest.7 The ability for ultrasound to be used as a primary prognostic tool in the neck to help dictate whether elective neck dissection is necessary has been investigated but has not changed the treatment paradigm in the N0 head and neck patient.
Benign Neoplasms of the Neck
Vascular Neoplasms
Pathology
Normal paraganglia contain two types of cells: type 1, chief cells or granular cells; and type 2, the supporting or sustentacular cells. Type 1 cells contain dense-core granules filled with catecholamines, a property that places them in the amine precursor and uptake decarboxylase (APUD) system. Type 2, or sustentacular cells, are elongated cells that closely resemble Schwann cells. Their function is not entirely clear. Tumors of paraganglia such as carotid body tumors contain both type 1 and 2 cells. Type 1 cells predominate and are arranged in an organized-nested pattern, known as Zellballen, surrounded by sustentacular cells in a fibrous stoma. These nests of Zellballen are illustrated in Figure 118-1. Type 1 chief cells tend to be polygonal shaped with abundant granular eosinophilic cytoplasm. They are peripherally surrounded by type 2 sustentacular cells that are difficult to identify by light microscopy and appear as spindle-shaped basophilic cells. Nuclear pleomorphism and cellular hyperchromatism are common in paragangliomas and should not be considered evidence of malignancy. Immunohistochemistry aids the diagnosis and differential diagnosis of these neoplasms. Type 1 cells stain positively with neuron-specific enolase, chromogranin A, and synaptophysin. Type 2 cells stain with S-100 and focally with glial fibrillary acidic protein.
Nomenclature
Other terms such as chemodectoma, glomus tumor, and nonchromaffin tumor are less accurate terms and should be avoided. Chemodectoma is an inaccurate term to describe all paragangliomas of the head and neck because the carotid body is the only known paraganglia of the head and neck that behaves as a chemoreceptor. The term glomus tumor more accurately describes benign cutaneous tumors arising from neuromyoarterial cells surrounding arteriovenous anastomoses. Designation as nonchromaffin tumor relates to histologic staining characteristics. An early histologic staining technique employing the chromaffin reaction failed to show the presence of catecholamines; paragangliomas were therefore described as nonchromaffin tumors. Newer techniques however have detected catecholamines in small quantities.8 The chromaffin reaction is a highly insensitive method on which to classify these tumors.9
Epidemiology
The most common paraganglioma of the head and neck is the carotid body tumor, followed by jugulotympanic paragangliomas and vagal paragangliomas. Other sites include the larynx, nasal cavity, orbit, trachea, aortic body, lung, and mediastinum. It has been estimated that paragangliomas comprise 1 in 30,000 head and neck tumors.10 However, the true incidence of paragangliomas may be unknown because previous reports have confused paragangliomas with neuroendocrine tumors.11 Further complicating an accurate estimate is the multicentricity of these tumors, particularly in familial paragangliomas.
Carotid Paragangliomas
History
The anatomist von Haller first described the carotid body in 1743; its function, however, was unknown at the time. Histologic studies of the carotid body revealed glandular acini, so the carotid body was renamed the carotid gland. Von Luschka first described a tumor of the carotid body in 1862. In 1880 Reigner performed the first resection of a carotid body tumor, but the patient did not survive. Six years later Maydl resected a carotid body tumor, and the patient survived but had postoperative hemiplegia and aphasia. In 1889 Albert was the first surgeon to resect successfully a carotid body without ligating the carotid vessels. The first successful removal of a carotid body tumor in the United States was reported by Scudder in 1903.12 The term paraganglion was first used by histologist Kohn in 1903 to describe the carotid body.13 This term was most appropriate because cells of the carotid body originate from the neural crest and migrate in close association with autonomic ganglion cells, hence the name “paraganglionic.” In 1950 Mulligan described in the dog neoplastic degeneration of the carotid body as chemodectoma because of the chemoreceptor function of the carotid body.14
Anatomy and Physiology
The carotid body is located in the adventitia of the posteromedial aspect of the bifurcation of the common carotid artery. The normal carotid body measures 3 to 5 mm in diameter but is often larger in persons living at higher altitudes. The average weight of the normal adult gland is 12 mg, with a wide range previously reported as 1 to 47 mg.15 During surgical removal, the typical finding is a small, reddish-brown to tan, ovoid structure attached to the carotid vessels at the bifurcation by Mayer’s ligament, through which the feeding vessels run, primarily from the external carotid artery. Blood flow and oxygen consumption of the carotid body, gram for gram, exceed those of the brain or thyroid gland.16 Sensory innervation is from Hering’s nerve, a branch of the glossopharyngeal nerve that originates approximately 1.5 cm distal to the jugular foramen.
Etiology
The etiology of paragangliomas appears to be multifactorial. Most paragangliomas are solitary. Multiple pheochromocytomas and paragangliomas are seen in familial syndromes, mainly multiple endocrine neoplasia types 2A and 2B. Other syndromes associated with paragangliomas are neurofibromatosis type 1 and von Hippel-Lindau disease, which is characterized by retinal angiomas and cerebellar hemangioblastomas. Carney’s triad demonstrates the association of paraganglioma, pulmonary chondroma, and gastric leiomyosarcoma.17
In addition to these associations, a syndrome of familial paragangliomas characterized by multiple paragangliomas, especially in the head and neck region, has been described and occurs in at least 10% of cases. The familial nature of carotid body tumors was first suggested by Chase in 1933 in his description of two sisters with carotid body tumors.18 Many developments have occurred recently in the characterization of the familial paraganglioma (PGL) syndromes including the genes and mutations involved and recommendations for the head and neck surgeon in managing these patients and their at-risk family members (Table 118-1). Genetic mutations responsible for the hereditary form of PGL have been identified in genes coding for succinate-dehydrogenase subunit D (SDHD), B (SDHB) and C (SDHC) genes, which map to chromosome 11, 1 and 1, respectively. Hereditary PGL syndrome has been classified genetically into four entities: PGL1, PGL2, PGL3, and PGL4. Germline mutations in SDHD, SDHB, and SDHC have been identified in PGL1, PGL4, and PGL3, respectively. The gene for PGL2 has not been identified.19 Individuals with hereditary paraganglioma syndrome have early onset of tumors and a higher frequency of bilateral and/or multiple tumors than do those with sporadic disease. Past reports suggested familial paraganglioma to be rare, although recent literature challenges this assertion and suggests the association of paragangliomas with germline genetic mutations is likely to be significantly higher, representing 17% of sporadic patients in one study19 and up to 28% to 40% in other studies.20–22
Table 118-1 Screening Recommendations for Carriers of SDHB, SDHD, or SDHC Gene Mutations*
| Annual physical examination and measurement of blood pressure |
| Annual levels of urinary catecholamines and metanephrines |
| Imaging of neck, thorax, abdomen and pelvis every 6-12 mo by computed tomography and/or magnetic resonance imaging |
* Starts in early teens. Succinate-dehydrogenase subunits D (SDHD), B (SDHB), and C (SDHC).
From Drucker AM, Houlden RL. A case of familial paraganglioma syndrome type 4 caused by a mutation in the SDHB gene. Nat Clin Pract Endocrinol Metab. 2006;2(12):706-712 and Isik C, Erem C, et al. Familial paraganglioma. Eur Arch Otorhinolaryngol. 2006;263(1):23-31.
PGL1 is caused by mutations in the D subunit of the succinate dehydrogenase (SDH) gene and is the most common inherited genetic abnormality in families with a history of paragangliomas. All three syndromes follow an autosomal pattern of inheritance, but the inheritance pattern for PGL1 is autosomal dominant modified by genomic imprinting. Genomic imprinting in paragangliomas was described by van der Mey and colleagues after reviewing data from 15 large Dutch pedigrees.23 The imprintable gene is transmitted in a mendelian manner, but the expression of the gene is determined by the sex of the transmitting parent. With paragangliomas, the gene results in the development of a tumor when it is paternally inherited. Offspring of male carriers were observed to demonstrate a 50% incidence of tumors, whereas children of female carriers never developed tumors. PGL1 is associated most commonly with head and neck tumors but also confers a risk for pheochromocytoma. It is estimated that 86% of individuals with a gene mutation will develop a tumor by age 50.24 Gene mutations in SDHC are rare, with few families identified to date.
Of further interest is the development of a paraganglioma syndrome in head and neck patients due to the inheritance of mutations in the SDHB gene. Although the PGLs associated with mutations in SDHB are less common than their D subunit counterpart, mutations in this gene confer a higher risk for the development of extra-adrenal, catecholamine-secreting, and often malignant pheochromocytomas, making identification of these individuals crucial in management of the at-risk family members to ensure that proper screening for pheochromocytoma and other paragangliomas takes place. Several cases of renal cell carcinoma in carriers of these mutations have also been reported, and one case of papillary thyroid cancer has been documented in the literature.24 The role of genetic counseling and testing in this population of patients cannot be overemphasized. Due to the apparent high rate of mutations in patients with a history consistent with sporadic paragangliomas, consideration of genetic counseling and testing should be given to every patient with a carotid body tumor or other paragangliomas.
Clinical Presentation and Diagnosis
The characteristic feature of carotid body tumors is slow growth rate, which is reflected clinically by the delay between the first symptoms and the diagnosis, which averages between 4 and 7 years. A carotid body tumor usually presents as a lateral cervical mass, which is mobile laterally but less mobile in the cranio-caudal direction because of its adherence to the carotid arteries. This physical finding has been called a positive Fontaine sign.25 Alternatively, a carotid body tumor may present as a parapharyngeal mass. Many carotid body tumors are pulsatile by transmission from the carotid vessels or, less commonly, expand themselves, reflecting their extreme intrinsic vascularity. Sometimes a bruit may be heard by auscultation, but it can disappear with carotid compression. The consistency varies from soft and elastic to firm and these tumors are generally nontender. As they enlarge, progressive symptoms of dysphagia, odynophagia, hoarseness, and other cranial nerve (IX-XII) deficits appear. Carotid sinus syndrome syncope has been described in association with carotid body tumors.26 The syndrome refers to a loss of consciousness accompanied with a reflex bradycardia and hypertension. Inciting stimuli include spontaneous movement of the head or following digital pressure applied to the tumor. Rarely, paraganglioma of the head and neck may present as a functional neuropeptide secreting tumor.
The capacity for catecholamine synthesis in head and neck paragangliomas, however, does not translate immediately to clinical findings. Although all paragangliomas have neurosecretory granules, only 1% to 3% are considered functional.27 Glenner and coworkers first described a functional carotid body tumor secreting norepinephrine in 1962.9 Patients should be asked about signs and symptoms indicating elevated catecholamines. Complaints of headaches, palpitations, flushing, and perspiration should be evaluated. In these patients a 24-hour urine collection is examined for norepinephrine and its metabolites including vanillylmandelic acid (VMA) and normetanephrine. Alternatively, plasma metanephrine may be assessed. Excess epinephrine or metanephrine should prompt suspicion of an adrenal pheochromocytoma because head and neck paraganglioma lack the enzyme to convert norepinephrine to epinephrine (phenylethanolamine-N-methyltransferase). An abdominal CT scan should be performed to rule out a concomitant adrenal pheochromocytoma. α- and β-Adrenergic blocking is undertaken if a tumor is found to be functional preoperatively. This decreases the risk from sudden catecholamine release that may occur with tumor manipulation in surgery. Routine screening for urinary metanephrines and VMA and serum catecholamines is only indicated for multiple or familial paragangliomas or in the presence of catecholamine-related symptoms.28
Malignancy
Cellular criteria for malignancy have not been established. Harrington and Dockerty29 attempted to classify malignant tumors of the carotid body. Criteria for malignancy included mitoses with giant cells, nuclear pleomorphism, and capsular invasion. Using these criteria, 50% of the 20 tumors studied would be considered malignant. Batsakis30 concurred that increased mitotic rate and capsular invasion should not be considered as determinants of malignancy. Other authors have hypothesized that all carotid body tumors demonstrate some degree of capsular invasion.31 Malignancy is determined by metastasis, which must be proven with biopsy, because paragangliomas may exhibit multicentricity. There are no histologic criteria for malignancy. In fact, previous reports have described metastatic carotid body tumors without mitoses.32 The diagnosis of malignancy should be made by evidence of spread to regional lymph nodes or distant sites33,34 (most common being lung and bones).
Malignant paragangliomas have been reported in 6% of carotid body paragangliomas by Batsakis.30 Accurate 5-year survival rates are not available because of the low malignancy rate of an uncommon tumor. Data from the National Cancer Data Base35 suggest an overall 5-year survival rate of 60%. Distant metastases had a worse prognosis with a 5-year survival rate of 11.8%, while those with regional spread of disease fared much better with a 5-year survival rate of 78%.
Imaging Studies
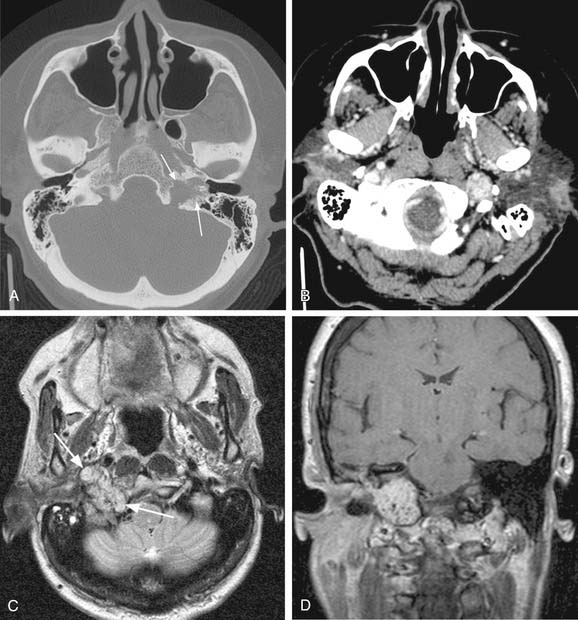
MRI with gadolinium may be the most useful imaging study for evaluating carotid body tumors (see Fig. 118-1A and B) because it offers superior soft tissue contrast without the need for ionizing radiation compared with CT scanning. MRI is sensitive for tumors as small as 0.8 cm.36 Paragangliomas larger than 2 cm in diameter typically demonstrate on T2-weighted images internal flow voids, dark lines, and dots that correspond to vascular structures. This is, however, not always present with carotid paragangliomas. Carotid body tumors demonstrate a characteristic lyre sign characterized as a bowing and displacing of the internal and external carotid arteries as shown in Figure 118-1B and C. Radiographic evaluation should be sufficient to make the diagnosis of carotid body tumor.
Carotid angiography has been replaced by MRI (including MR angiography, see Fig. 118-1C), but catheter angiography remains useful when preoperative embolization is necessary (see Fig. 118-1D). The use of preoperative embolization is controversial, but many authors have recommended its use for large tumors to decrease blood loss.37 Preoperative embolization may also be used in those rare instances where a malignant carotid body may be suspected. If preoperative embolization is planned, surgery should be performed 24 to 48 hours following in order to avoid revascularization, edema, or local inflammation. Additionally, angiography with temporary balloon occlusion using clinical and electroencephalographic monitoring, combined with brain CT perfusion scanning, can provide specificity as to the tolerance of collateral cerebral circulation across the circle of Willis in select cases.
The high density of somatostatin receptors in paragangliomas provides for newer functional nuclear medicine imaging techniques including metaiodobenzylguanidine (MIBG) scanning and octreotide scanning. MIBG scanning uses I-131 labeled tracer, which is concentrated in intracellular storage vesicles of paragangliomas.38 Octreotide scanning uses Indium-111 labeled somatostatin analog octreotide to diagnose primary tumors of the APUD system, as well as their metastases.39 These functional imaging studies have been recommended as a possible screening test for familial paragangliomas for patients at risk.40 They also allow the detection of additional tumors when malignant paraganglioma is suspected.41
Classification
Although not universally adopted in the literature on carotid body tumors, a classification system has been previously proposed for carotid body tumors. In 1971, while a surgery resident at the Mayo Clinic, Shamblin and coworkers described a classification system used to grade the difficulty of resection in carotid body tumors.42 Group I tumors were defined as localized, relatively small, and minimally attached to the carotid vessels. Surgical excision was described as carried out without difficulty in this group. Group II encompassed tumors adherent to or partially surrounding the vessels, with moderate arterial attachment. These tumors were described as amenable to careful surgical removal. Group III carotid body tumors completely encased the carotids. Shamblin and his colleagues recommended approaching these tumors with great care and with consideration for vessel replacement.
Surgery
In a report from Memorial Sloan-Kettering Cancer Center, Lack and colleagues discussed 39 of 43 patients with carotid body tumors treated surgically; one patient received definitive radiation therapy; and 3 others were observed but not treated.43 In this cohort of patients, 24 of 39 patients were free of disease following surgery, at an average follow-up interval of 12 years (6 months to 38 years). Local recurrence occurred in 4 of 39 patients (10%). Regional or distant metastases occurred in 4 of 39 patients (10%). All four of these patients were dead of disease within 6 years.
The incidence of permanent cranial nerve impairment as a complication of surgery has been reported in the literature to occur in approximately 20% of cases. In this report by Lack and colleagues,43 cranial nerve sacrifice of the vagus or hypoglossal nerve was necessary in 15% (6 of 39) of patients. An additional patient developed Horner’s syndrome postoperatively. Although not quantified, the superior laryngeal nerve supplying sensory innervation to the larynx and motor innervation to the cricothyroid muscle may be the most frequently injured nerve during carotid body resection. Paralysis of one superior laryngeal nerve may result in some degree of aspiration, although isolated palsy of one superior laryngeal nerve typically requires no additional rehabilitation. Furthermore, denervation of one cricothyroid muscle may result in pitch changes in singers, but changes in voice may not be perceptible. Injury to the cervical sympathetic chain will result in Horner’s syndrome, with ipsilateral ptosis, miosis, and anhidrosis. Netterville and colleagues have described first bite syndrome resulting from injury to the cervical sympathetic chain, resulting in loss of sympathetic input to the parotid gland.36 Patients with first bite syndrome complain of severe cramping in the parotid area when they take the first bite of food, particularly with the first meal of the day. The pain generally subsides over the next several bites but is more intense with strong sialogogues such as tart or bitter foods. The physiologic mechanism behind first bite syndrome is likely due to denervation supersensitivity of the sympathetic receptors that control the myoepithelial cells in the parotid gland. With oral intake, parasympathetic neurotransmitters are released and cross-stimulation of the sympathetic receptors causes a supramaximal response of the myoepithelial cells. Treatment includes restriction to bland foods and oral carbamazepine (100 to 200 mg twice daily) for those with severe pain.
Anand and colleagues44 reviewed 1181 published cases of carotid body tumors treated with surgical resection. Internal carotid artery injury was identified in 23% of cases (275 cases), with an overall occurrence of central nervous system (CNS) complications of 26%. This subcategory of internal carotid artery injury was further examined. In 23% of cases (62 cases), internal carotid artery repair was accomplished with simply suture or patch repair, with a CNS complication rate of 3%. The internal carotid artery was reconstructed in 45% (125 cases), with a CNS complication rate of 10% and a mortality rate of 2%. It was necessary to ligate the internal carotid artery in 32% (89 cases), resulting in a CNS complication rate of 66% and a mortality rate of 46%.
Controversy over Surgery for Multicentric Tumors
Another recently described and rarely recognized problem following bilateral carotid body excision is baroreflex failure syndrome.36 The clinical manifestations are caused by bilateral denervation of the carotid sinus. The carotid sinus is situated in the adventitia of the carotid bulb and serves as a baroreceptor to decrease systemic blood pressure. Bilateral baroreceptor dysfunction causes unopposed sympathetic outflow, resulting in marked fluctuations in blood pressure and a sustained tachycardia postoperatively. Over time, compensation occurs, but it is variable and unpredictable. Compensation may occur by baroreceptor fibers in the aorta or through neural regrowth at the carotid sinus. Therapy consists of sodium nitroprusside in the early recovery phase to prevent excessive hypertension. Long-term control may be accomplished using oral antihypertensives such as clonidine or phenoxybenzamine. Clonidine is an α-2 agonist, resulting in decreased release of norepinephrine into synaptic clefts, and stimulation of parasympathetic outflow, slowing the heart rate. Phenoxybenzamine is an α-1 and α-2 antagonist that decreases peripheral resistance and increases cardiac output.
Radiation Therapy
Radiation oncologists at the University of Florida have described effective local control of 23 carotid and vagal paragangliomas using definitive radiotherapy.45 In their study, 15 patients with 23 carotid or vagal paragangliomas were treated with radiotherapy between 1981 and 1995. Nineteen tumors were treated with radiation therapy alone, and four were treated with surgery and postoperative radiation therapy. For benign tumors, total doses ranged from 35 to 48.5 Gy. The two malignant tumors received 64.8 and 70 Gy, respectively. Follow-up ranged from 1.5 to 10 years. Local control was achieved in 96% of tumors at 5 years. Five-year disease-specific survival was 89%, with one patient dead of locally recurrent disease 5 years after radiation therapy. This patient had been previously treated with surgery and radiation therapy before treatment by the authors. Another patient died of atherosclerotic disease 13.5 years after radiation therapy. There were no patients with regional or distant failure following treatment. Complications reported by these authors included one patient experiencing a delayed transient CNS syndrome. No other complications were reported.
Valdagni and Amichetti reported 7 patients with 13 carotid body tumors treated with radiation therapy between 1968 and 1987.46 Treatment consisted of radiotherapy alone for 10 tumors and surgery plus radiation therapy for 3 tumors. Total doses ranged from 46 to 60 Gy at 1.8 to 2.5 Gy per fraction. Follow-up ranged from 1 to 19 years. Local control was achieved in all patients. Acute side effects were minimal. No short or long-term toxicities were reported.
Even proponents of radiation therapy for carotid body tumors concur with surgical resection as the preferred modality of treatment for most lesions.47 Definitive radiation therapy may be reserved for patients who are poor surgical candidates due to a debilitated medical condition and for locally advanced tumors where anticipated postoperative morbidity may preclude consideration for surgical resection. Adjuvant radiation therapy may be considered following surgery for malignant carotid body tumors for locoregional control.
Observation
Using sequential radiologic imaging, Jansen and colleagues48 have estimated the median tumor doubling time for 20 carotid body tumors to be 7.1 years including 12 cases without detectable growth. Farr has estimated a growth rate for carotid body tumors, plotting tumor size against years of symptom duration, of 2 cm in 5 years.49 A scan-and-wait policy may be considered for those patients who are not suitable candidates for surgery or radiation therapy. This highly select group of patients includes those individuals whose medical condition is so poor that both surgery and radiation therapy are contraindicated, or those patients of such advanced age that the carotid body tumor may have minimal impact on their life expectancy or quality of life. This group may also include those patients with malignant carotid body tumors with distant metastases where locoregional treatment would be only palliative in intent.
Vagal Paragangliomas
Vagal paragangliomas are tumors derived from paraganglionic tissue associated with one of the ganglia of the vagus nerve.50 Vagal paragangliomas most commonly arise from the inferior vagal ganglion, also referred to as the nodose ganglion. Tumors arising from the superior vagal ganglion, or jugular ganglion, may be dumbbell-shaped and may extend from the neck intracranially through the jugular foramen.
Clinical Presentation and Diagnosis
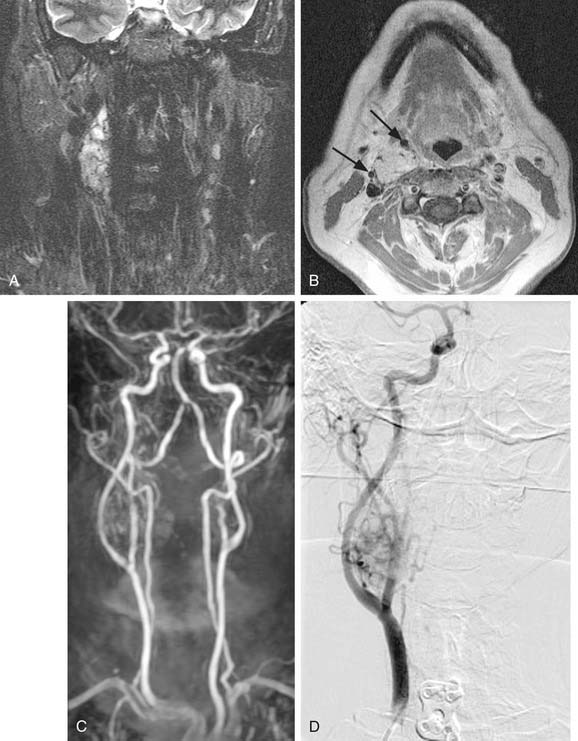
Vagal paragangliomas, like carotid body tumors, may present as a palpable neck mass that is more mobile in a lateral direction than a craniocaudal orientation. Paralysis of the ipsilateral true vocal cord or Horner’s syndrome, from involvement of the ipsilateral sympathetic chain, may be present as the tumor grows in size. True vocal cord paralysis may result in hoarseness with or without aspiration of liquids from glottic incompetence. Large tumors arising from the jugular ganglion may be associated with cranial neuropathies of IX, XI, and XII. Diagnostic imaging studies described previously in the chapter may demonstrate anterior displacement of the carotid artery from the tumor present in the posterior carotid sheath. Unlike carotid body tumors, the internal and external carotid arteries do not manifest a splayed configuration in vagal paragangliomas (Fig. 118-2).