Molecular Diagnostics of Ocular Infectious Disease: Polymerase Chain Reaction Diagnostics of Ophthalmic Disease
Elma Kim
Russell N. Van Gelder
In this chapter, we review the biochemistry, indications, applications, and limitations of the polymerase chain reaction (PCR) for the diagnosis of ocular disease. Although PCR is also used in the genetics laboratory for identifying specific mutations, we do not consider that application but restrict our discussion to the use of PCR in the diagnosis of the infectious diseases of the eye.
HISTORY AND BIOCHEMISTRY OF THE POLYMERASE CHAIN REACTION
The modern PCR was first described in 1985.1 In the 20 years since the first publication, the paper describing the modern version of PCR2 has been cited more than 13,000 times, and more than 190,000 papers have been published using this technique.
PCR is essentially an in vitro replication of bacterial DNA replication, stripped of all inessential components (Fig. 1). The necessary ingredients are the DNA template (i.e., from a pathogen residing in a biopsy specimen), a pair of 20 to 30 nucleotide oligonucleotide primers specific for the targeted DNA sequence (i.e., a sequence from a viral genome), nucleotide triphosphates (the building blocks for DNA), a thermostable DNA polymerase to catalyze replication, appropriate salts and buffers, and a thermal cycling machine. Once combined, the thermal cycler creates conditions for the three steps of PCR. Each step usually takes between 30 seconds and 2 minutes. The process begins with the denaturation step (typically at approximately 94°C), which separates the double-stranded template DNA into single strands. The temperature is then lowered to the annealing temperature (specific to each pair of primers, typically 45°C to 65°C), which allows the primers to optimally bind to their target sequences. After annealing, the temperature is elevated to an optimal temperature for thermostable DNA polymerase activity (typically between 72°C and 74°C). DNA polymerases capable of functioning at such high temperatures are isolated from thermophilic bacteria (such as those living in geothermal pools). At this temperature, the polymerase catalyzes synthesis of DNA complementary to the template at a rate of approximately 1000 bases per minute; thus a 500 base pair (bp) target may require 30 seconds of extension time. After extension, the temperature is again raised to denature the template DNA from the newly formed complementary strand of DNA. Thus, at the end of the first cycle, the number of target DNA strands has doubled. Both the original template and the new complement strand can then become templates for another round of amplification. PCR is an exponential process; theoretically, after 35 cycles (standard for many protocols), more than 35 billion copies of the original template DNA will have been synthesized. Starting with a single virion (whose DNA weighs 1.6 × 10-16 g), after 35 cycles nearly 20 ng (2.0 × 10-8 g) of 500 bp product will be produced. This quantity of DNA is adequate for further analysis.
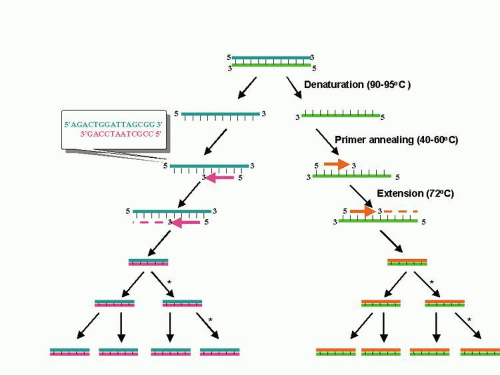 Fig. 1. Schematic of the polymerase chain reaction. Each cycle of the polymerase chain reaction (PCR) includes heat denaturation of the double-stranded DNA template into single strands, annealing of an oligonucleotide primer complementary to sequence being amplified, and extension of the annealed primer using a thermostable DNA polymerase and nucleotide triphosphates. Repeated cycles results in exponential, sequence specific amplification of the original target. |
The most commonly used method for detection of the PCR product is electrophoresis in an ethidium bromide-stained agarose gel (Fig. 2). The limit of detection in such a gel is approximately 10 ng of double-stranded DNA. Given the calculations above, PCR should theoretically be able to detect a single genome in a given sample. However, because of imperfect amplification efficiency, maximal sensitivity is generally on the order of 10 genomes. PCR products may also be detected in other ways, using more sensitive dyes such as fluorescent SYBR Green®. This dye may be used in a special variant of PCR (real-time PCR), which allows quantification of product (see later). Sensitivity for detection of PCR products can be improved by use of radioactivity and autoradiography, but these techniques are seldom applied in clinical practice.
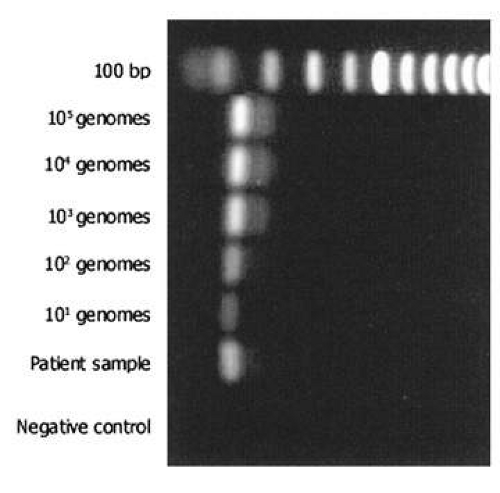 Fig. 2. Typical ethidium bromide-stained gel electrophoresis after polymerase chain reaction (PCR) from ocular specimen. First lane shows molecular weight marker. Positive controls are a dilution series of purified herpes simplex DNA, ranging from 105 genomes to 101 genomes. Patient sample is positive and appears to contain between 102 and 103 genomes. Negative control includes all reaction components but substitutes water for patient sample. |
Although detection of a band of the correct size on electrophoresis is generally considered indicative of a positive result, PCR products may be more specifically identified in a number of ways. Restriction endonucleases can digest the PCR product into sequence-specific fragments, the pattern of which, on electrophoresis, forms a distinct fingerprint. Alternatively, hybridization methods allow identification by binding the PCR product to a nylon or nitrocellulose filter paper and then incubating the nylon with a labeled DNA or RNA probe that binds to a specific DNA sequence. The gold standard for demonstration of a specific identity for a PCR product is DNA sequencing, which can be performed either directly from the PCR product or after cloning of that fragment into a plasmid vector.
SPECIMEN ACQUISITION FOR DIAGNOSTIC POLYMERASE CHAIN REACTION
Ophthalmic samples for PCR are typically collected in one of three ways: swab samplings of the cornea, conjunctiva, or adnexa; anterior chamber paracentesis; or vitreous biopsy. The type of biopsy taken is guided by disease suspicion, media opacity, structural parameters of the eye, coexistence of pathologic conditions, and the experience of the ophthalmologist. In general, the sample should be taken at the site closest to pathology. For example, vitreous sampling has higher yield than aqueous sampling for ocular toxoplasmosis (although for viral retinitis, both anterior chamber and vitreous biopsy have high yield, likely because of higher infectious burden and smaller and more diffusible pathogen). All samples should be collected aseptically and placed into a sterile container. For a given PCR reaction, 5 to 10 μL of sample is sufficient. However, to allow retesting or testing for multiple pathogens, larger volumes are necessary. Approximately 50 μL from an anterior chamber paracentesis is generally sufficient and should be immediately capped in a 1-mL sterile syringe. For vitrectomy samples, approximately 50 to 100 μL of a dry (i.e., infusion-off) aspirate is collected in a sterile tube and capped upon collection. While PCR can theoretically be performed from vitrectomy cassette material, the extreme dilution of sample by this technique leads to a high incidence of false-negative results.
For external disease, placement of the swab depends on disease presentation. For conjunctivitis, the swab is placed on the conjunctiva without touching the patient’s external skin. For a corneal ulcer, the swab should be placed along the periphery of the ulcer, in an active-appearing region. Once the sample is collected, the swab should be placed in a sterile tube containing 0.1 mL of balanced salt solution. In the laboratory, the swab is “milked” and subsequently prepared for PCR.
Once obtained, samples should be placed on ice and, at the earliest opportunity, quick-frozen on dry ice or in liquid nitrogen. The sample should remain frozen because freeze-thaw cycles are potentially deleterious to the sample, potentially degrading nucleic acids (primarily RNA). Swab samples should be stored at -80°C until laboratory processing.
All samples, regardless of source, must be processed to eliminate potential PCR inhibitors.3 Boiling the sample for 10 to 15 minutes is an effective way to remove most inhibitors and to release nucleic acids from relatively acellular sources (such as aqueous or vitreous). Alternatively, inhibitors can be removed by a commercially available DNA purification kit. Such purification is important when analyzing highly cellular samples such as corneal or scleral biopsies. Importantly, if the sample is to be tested for intraocular antibodies (i.e., Goldmann-Witmer testing4), the laboratory must be notified because boiling irreversibly denatures antibodies from the sample. In these cases, the sample must be aliquoted for separate PCR and antibody testing.
It is essential that both negative controls (water or balanced saline in lieu of patient sample) and positive controls (a quantified sample of known pathogen DNA) be analyzed simultaneously with each PCR sample. Laboratory contamination is a potential cause of false-positive results in PCR; because PCR products are also potential targets for amplification, great care must be taken to avoid contamination of reagents with previous products. The use of consumable supplies and reagents, careful techniques, physical separation of reagents and controls, and routine inclusion of negative controls are necessary. Deoxyuracil may be substituted in PCR for the base thymidine; this base can be deglycosylated with uracil DNA glycosylase prior to the PCR reaction to degrade any product that could potentially serve as template.5
VARIANTS OF POLYMERASE CHAIN REACTION
NESTED POLYMERASE CHAIN REACTION
To increase sensitivity further, nested PCR may be used. Nested PCR is a second round of PCR that uses first-round amplification PCR products as the template DNA. The set of primers used in a nested PCR are internal to the primer pair used in the original PCR (Fig. 3). Nested PCR can have single genome sensitivity; it can also be utilized to improve specificity for detection of a specific pathogen. However, the large number of amplification cycles used in nested PCR (sometimes more than 60) can markedly increase the probability of false positive results. We generally avoid use of nested PCR for routine diagnostic applications.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


