Chapter 23 Mechanisms of ER Stress in Retinal Disease
The endoplasmic reticulum
As its name suggests, the ER is a membrane-bound labyrinth of tubes and sacs, where virtually all plasma membrane and secreted proteins begin their progression to the cellular surface. The ER is the largest membrane compartment within eukaryotic cells, frequently comprising more than half of the total membrane composition. The ER has a number of major physiological functions.1 It plays a key role in lipid and protein biosynthesis; it functions as a Ca2+ storage organelle. Importantly, it is the site where most secretory and transmembrane proteins fold into their native conformation. When proteins enter the ER, they may undergo formation of disulfide bonds through the action of folding enzymes such as protein disulfide isomerase (PDI).2 Proteins that are processed through the ER frequently undergo glycosylation, a posttranslational modification involving the addition of asparagine-linked oligosaccharides. There are two known chaperone systems in the ER, the calnexin/calreticulin system and binding protein/glucose-regulated protein 78 (BiP/GRP78). The calnexin/calreticulin system is particularly relevant to folding of glycoproteins, although some calnexin/calreticulin substrates can also bind BiP/GRP78. Protein folding is essential for protein function; therefore, sophisticated mechanisms have evolved to ensure that proper folding occurs or that irreversibly misfolded proteins are eliminated. Quality control is an ER surveillance mechanism to permit only properly folded proteins to exit the ER en route to other organelles.
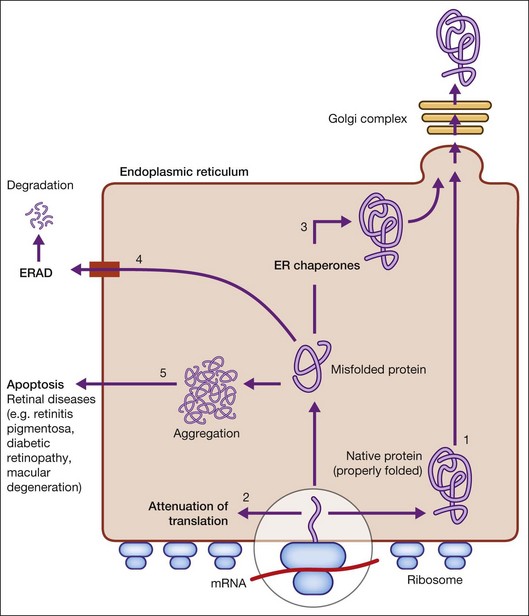
When there is perturbation of ER function (such as inhibition of glycosylation or disulfide bond formation, disruption of Ca2+ homeostasis, hypoxia, and infection), unfolded or misfolded proteins accumulate. This situation, termed ER stress, is defined as an imbalance between the cellular demand for ER function and ER capacity.2–4 When the influx of nascent unfolded polypeptides exceeds the folding and processing capacity of the ER, the normal physiological state of the ER is perturbed, activating signaling pathways, termed the ER stress response or the unfolded protein response (UPR), to return the ER to its normal physiological state.2–4 The possible fates for a protein in the ER are shown in Figure 23.1 and include: (1) proper folding and exit through the ER; or in the case of misfolded proteins: (2) translational attenuation whereby protein synthesis halts temporarily to prevent accumulation of unfolded proteins; (3) transcriptional induction of ER chaperone genes to increase protein folding capacity; (4) transcriptional induction of ER-associated degradation (ERAD) activity. If these strategies fail and protein aggregation is excessive, apoptosis (5) is induced to dispose of cells injured by ER stress, thereby ensuring survival of the organism. Recent studies implicate dysregulation of the UPR in neurodegenerative diseases, including retinal diseases.
ER stress and UPR signaling
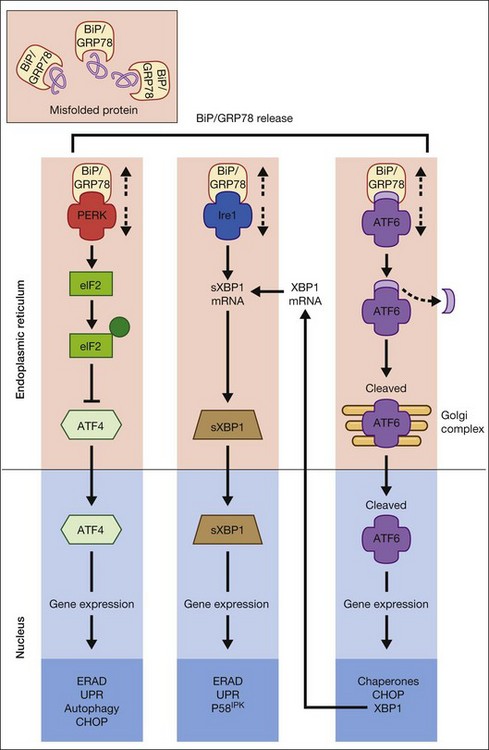
The UPR is a complex cellular signaling pathway that attempts to restore cellular homeostasis, but, in the face of prolonged ER stress, pathways may be activated that lead to cell death.5 The UPR involves three signaling pathways governed by three integral ER membrane proteins: PERK (protein kinase-like ER kinase, pancreatic ER eukaryotic translation initiation factor (eIF)-2a kinase), IRE-1 (inositol-requiring protein 1), and ATF6 (activating transcription factor 6). Research related to ER stress and retinal disease has focused on investigations of BiP/GRP78 and these downstream effector proteins.
Binding protein/glucose-regulated protein 78
BiP/GRP78 is a calcium-dependent resident ER protein that facilitates the transfer of proteins into the ER through the ER translocator.2,3,6 It is a heat shock protein with a molecular weight of 78 kDa. As a key ER stress regulatory protein, it binds to exposed hydrophobic amino acid sequences of polypeptides that would normally be buried in the interior of correctly folded proteins. Under nonstress conditions, BiP/GRP78 binds to the luminal domains PERK, IRE1, and ATF6, maintaining them in an inactive state (Fig. 23.2). When misfolded proteins accumulate, BiP/GRP78 preferentially binds to these misfolded proteins and dissociates from the UPR sensor proteins allowing their activation. BiP/GRP78 and other UPR gene targets such as GRP94 and calreticulin contain an ER stress response element (ERSE, CCAAT(N9)CCACG) that is required and sufficient to activate the UPR. The absolute requirement for BiP/GRP78 for survival is underscored by evidence that absence of BiP/GRP78 results in perimplantation embryonic lethality in mice.7 Upregulation of BiP gene expression is considered by some investigators as a marker for ER stress induction.
PERK
The signaling events mediated by PERK represent the most immediate response to ER stress in metazoan cells. PERK is an ER-associated transmembrane serine/threonine protein kinase. When unfolded proteins accumulate in the ER lumen, BiP/GRP78 dissociates from PERK, which then dimerizes and is subsequently autophosphorylated, triggering phosphorylation of eukaryotic translation initiation factor 2 on the alpha subunit (eIF2α). Phosphorylation of eIF2α attenuates mRNA translation, thereby preventing influx of newly synthesized polypeptides into the ER lumen of the stressed cell.6 Interestingly, although phosphorylation of eIF2α inhibits translation initiation in general, it is required for selective translation of several mRNAs such as ATF4. Activation of ATF4 can increase levels of chaperones, restore cellular redox homeostasis, and help the ER to fold proteins or degrade them. Lack of the gene encoding PERK is not lethal, but does result in increased hypersensitivity to ER stress.8 It has been reported that activation of PERK induces transcription of ~1/3 of UPR-dependent genes.9
IRE1
IRE1 is a 100-kDa bifunctional protein with kinase and endoribonuclease (RNase) activity. It was the first component in the UPR pathway to be identified10 and is evolutionarily the oldest branch of the UPR. Under nonstress conditions, the protein kinase is maintained in a monomeric form through its interactions with BiP/GRP78. IRE1 can bind members of the tumor necrosis factor (TNF) receptor family and can activate protein kinases that are implicated in immunity, inflammation, and apoptosis. Under conditions of ER stress, when unfolded proteins are accumulating, IRE1 is released from BiP/GRP78, dimerizes, and autophosphorylates to activate its RNase activity.2–4 In mammals, there are two forms of the protein, IRE1α and IRE1β. IRE1α is expressed in most cells and tissues (including retina), while IRE1β is primarily in intestinal epithelial cells. Upon activation of the UPR, IRE1 RNase activity initiates removal of a 26-nucleotide intron from X-box binding protein (XBP1) mRNA. Spliced XBP1 is a transcriptional activator that activates many UPR target genes through its interaction with ERSE and it can activate genes required for ER-associated degradation (ERAD). IRE1 may be a focal point for the BCL-2 family of proteins that regulates cell death. BCL-2-associated X protein (BAX) and BCL-2 antagonist/killer (BAK) interact physically with IRE1α, modulating the UPR. If either IRE1 or XBP1 is absent in mouse models, the result is embryonic lethality.
ATF6
ATF6α and ATF6β are bZIP family transcription factors. In the absence of ER stress, BiP/GRP78 binds to the luminal domain of ATF6, tethering it to the ER membrane. When unfolded proteins accumulate, BiP/GRP78 releases ATF6, which then translocates to the Golgi apparatus by vesicular transport.3,6 Unlike PERK and IRE1α, ATF6α and ATF6β do not undergo oligomerization, rather in the Golgi ATF6 is cleaved by proteases and the resultant cytoplasmic portion translocates to the nucleus, where it (like XBP1) binds to ERSE to activate transcription of ER chaperone genes such as BiP/GRP78, GRP94, and calreticulin. Thus, ATF6 activation can increase ER chaperone activity. If both ATF6α and ATF6β are deleted in mouse, the result is early embryonic lethality.
ER-associated degradation
The ER employs ERAD to clear aggregated, misfolded, or unassembled proteins. Target proteins are selected through the ER quality control system, retrotranslocated to the cytosol, and degraded by the ubiquitin proteosome system. The four steps associated with ERAD are recognition, retrotranslocation, ubiquitination, and degradation.3 In the recognition step, glycosylated proteins are bound to ER degradation-enhancing α-mannosidase-like protein that can discriminate folded from unfolded proteins. Misfolded proteins destined for the retrotranslocation machinery associate with PDI and BiP/GRP78 to cleave disulfide bonds and to unfold the partially folded protein. Proteins are translocated to the cytoplasm where they undergo ubiquitination by the E1–E2–E3 ubiquitin system. The proteins are then deglycosylated and degraded by the proteosome.
Retinal diseases associated with ER stress
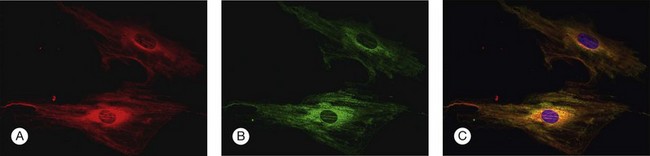
To investigate the role of ER stress genes and proteins in retina, a number of research tools (e.g., antibodies, molecular probes) are available commercially or have been developed by individual laboratories. Figure 23.3 shows immunocytochemical studies performed in freshly isolated mouse retinal Müller cells to detect two ER proteins. PDI is a known ER-resident protein and hence an excellent marker of this organelle (Fig. 23.3A). In this dual-label experiment, a second antibody was used to detect BiP/GRP78, the key ER stress-regulatory protein (Fig. 23.3B). The final panel shows the merged image of PDI and BiP/GRP78; there is considerable overlap in the expression of these two proteins (Fig. 23.3C). Immunodetection methods, gene expression analysis, and elegant genetic manipulation methods have formed the basis of investigations of the ER stress genes/proteins in various retinal diseases.
Retinitis pigmentosa and other photoreceptor dystrophies
RP is an inherited retinal dystrophy in which loss of photoreceptors leads to progressive vision loss. The prevalence of nonsyndromic RP is ~1/3500–4000. The most common form of RP is a rod–cone dystrophy, characterized initially by night blindness, followed by progressive loss in the peripheral visual field in daylight, eventually leading to blindness after several decades.11 To date mutations in over 40 genes have been implicated in RP.12 In some cases, the genes are specific to photoreceptor cells, including rhodopsin, rod cGMP phosphodiesterase, peripherin, and rod outer-segment membrane protein-1, while others are expressed in retinal and nonretinal cells.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


