Chapter 112 Management of Combined Inflammatory and Rhegmatogenous Retinal Detachment
Introduction
Retinal detachments (RD) occurring in the context of ocular inflammation are a particular challenge both to the vitreoretinal surgeon and to the uveitis specialist. Most often they occur during or following an episode of intraocular infection and are most commonly seen in eyes having suffered from a viral retinitis. When they are not related to ocular infection, their diagnostic and therapeutic approach may be difficult, since different forms of retinal involvement suppose different treatments and prognosis. Serous retinal detachment (SRD) is the form most commonly associated with active, purely inflammatory conditions and, often, its presence helps to establish the diagnosis, as in the case of Vogt–Koyanagi–Harada disease (see Chapter 75, Vogt–Koyanagi–Harada disease). Not infrequently, it is misdiagnosed for a rhegmatogenous retinal detachment (RRD). However, the management of SRD is entirely pharmacologic, aimed at controlling the intraocular inflammation, and not surgical. The visual prognosis in most cases of SRD is excellent provided that the immunosuppressive agents have been introduced at a sufficient dose to lead to rapid resolution, and tapering is done slowly. RRD in the setting of active uveitis is thankfully rare, but its presence, even with modern vitreoretinal surgical techniques, results in a guarded visual outcome. Tractional retinal detachment (TRD) is possible in both infectious and noninfectious uveitis whenever vitreous inflammation/organization develops in an eye without posterior vitreous detachment (PVD) or when proliferative vitreoretinopathy (PVR) complicates the course of intraocular inflammation. Combined forms of RD are also possible; it is crucial to establish the exact mechanism of visual deterioration in every single case, allowing a better therapeutic approach.
Epidemiology
A systematic review of population-based studies on RRD published between 1970 and 2009 found a reported incidence between 6.3 and 17.9 for 100 000 people/year, with significant geographical variations.1 It is widely accepted that RRD occurrence varies with ethnicity and is strongly associated with increasing age, myopia, vitreoretinal degeneration and following cataract surgery.2,3 Uveitis itself has not been considered among the predisposing conditions to develop RRD. However, a retrospective study conducted in the Netherlands found a prevalence of RRD of 3.1% among 1387 uveitic patients, suggesting inflammation as an independent risk factor for its development.4
Not all types of uveitis carry the same risk of RRD; this complication is most commonly seen in posterior uveitis from infectious etiology, with the highest risk in viral retinitis cases. Acute retinal necrosis (ARN) (see Chapter 88, Acute retinal necrosis syndrome) evolves to RRD in more than 50% of cases, in a median time of 53 days after presentation.5 This risk is even higher for progressive outer retinal necrosis (PORN), in which almost 70% of affected eyes will develop RRD soon after presentation.6 For cytomegalovirus (CMV) retinitis (see Chapter 81, HIV-associated infections), even though the course of the disease has positively changed with the introduction of the HAART therapy, the rate of RRD remains as high as 2.3/100 eye-years.7
Parasitic infections can lead to RD when a choroidal process extends to the retina and vitreous or when a retinal process causes overlying vitreous to condense and contract. Toxoplasmic retinochoroiditis (see Chapter 85, Ocular toxoplasmosis) presents a risk of RRD of 3.5–6%.8–10 In the subset of patients addressed for vitreoretinal surgery, Adan et al. found 53.3% of cases of RD, most of them with pure RRD, but also some cases of combined tractional and RRD.11 Exudative detachments are rarely associated with ocular toxoplasmosis but have been cited in the literature.12,13 Vision-threatening features in ocular toxocariasis (OT) (see Chapter 86, Helminthic disease) are mainly severe vitritis, cystoid macular edema and tractional detachment of the macula. A retrospective series on OT found a prevalence of macular traction of about 30%, regardless of the site where the choroidal granuloma was located. In peripheral lesions with retinal traction, TRD occurs in about 40% of cases.4,14
Among noninfectious uveitis, the risk of RRD is less than 1%. Most patients developing retinal tears and detachments appear to do so as a result of concomitant retinal pathology, often in areas unrelated to sites of inflammation. However, inflammatory processes affecting the vitreous base may present an exception to this rule. Pars planitis, when untreated for a prolonged period of time or if severe, is known to develop TRD15 and retinoschisis.16 The differentiation between true retinoschisis and TRD may sometimes be very difficult, especially for those lesions situated in the far periphery.17 Sarcoidosis is also known to develop various forms of RD (see Chapter 78, Sarcoidosis), though this complication is rare in this condition. Serous RD,18 RRD,19 retinal pigment epithelium (RPE) detachments,20 and even a rare case of necrotizing retinitis with subsequent RRD,21 were described in the context of ocular sarcoidosis, but mostly as isolated cases. Retinal tears and RRD are rarely found in relation to Behçet’s disease, even though detailed fundoscopy should be done periodically in these patients, mainly when vitreous inflammation is important. A case of RRD secondary to a macular hole in a young patient with Behçet uveitis22 has been reported, stressing the role of recurrent vitritis in the pathogenesis of this complication (see Chapter 77, Autoimmune retinopathies).
Pathophysiology
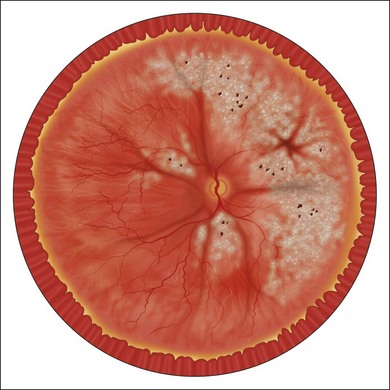
The nature of the inflammatory or infectious process determines the mechanism by which the detachment occurs, and provides an indication as to the means by which it can be resolved. Serous RD is usually the result of an immune-mediated assault to the choroid or the RPE. Its follows from loss of the tight junctions between retinal endothelial cells, leading to increased outward flow of fluid from retinal vasculature, and a failure of the compensatory pump function of RPE cells. Disruption of the normal oncotic gradient between the vitreous cavity and the choroid may also compromise passive transfer mechanisms responsible for a good portion of normal fluid flow to the choroid. Inflammatory mediators modulate vascular permeability and may have a role on the modulation of chloride receptors such as CLIC4 necessary for retinal attachment.23,24 Resolution of a serous detachment requires one to re-establish the status quo by inhibiting both humoral and cellular inflammatory mechanisms through appropriate systemic or local immunosuppression (Fig. 112.1).
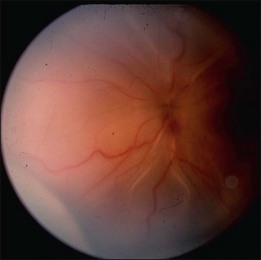
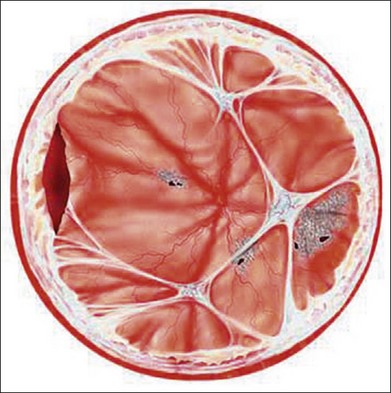
In the case of both RRD and TRD, vitreous body traction plays a predominant role. In RRD associated with toxoplasmosis, active toxoplasmic retinochoroiditis often precedes or is concurrent with the development of the RRD.8 Inflammation not only enhances vitreous liquefaction but also leads to cross-linking between collagen fibrils, particularly those close to the retinal interface, leading to the formation of an anomalous PVD and retinal tears.25,26 Vitreal strands between the optic disk and the ocular toxocariasis granuloma are present in nearly 50% of patients (Fig. 112.2). These strands are often the source of retinal traction and detachment. Contraction of the vitreous base, particularly seen in chronic longstanding uveitis, leads to the development of fibrous or fibrovascular bands, which can be very adherent to the underlying retina.26,27 Their removal can be quite challenging at the time of surgery (Fig. 112.3).
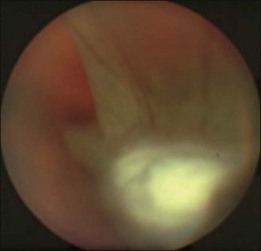
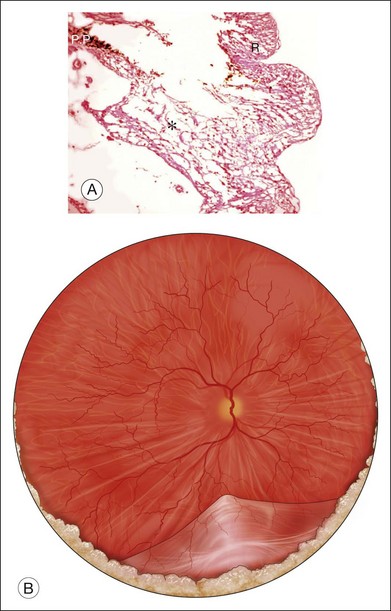
High levels of s-ICAM1 (type 1 soluble intercellular adhesion molecule) are found in vitreous from uveitic eyes complicated by RD similar to what has been observed in cases of proliferative vitreoretinopathy (PVR) where it is felt to perpetuate a cytokine-mediated vascular reaction which may contribute to PVR formation.28–30 It correlates positively with vitreous levels of TNF-α (tumor necrosis factor-alpha), a cytokine with a key role in the initiation of the inflammatory cascade and the pathogenesis of uveitis.31
Herpetic retinal infections (CMV, HSV, VZV) are characterized by retinal necrosis leaving in its wake a diaphanous glial membrane (Fig. 112.4). Depending on the interplay between the virus and the host immune system, it is associated with moderate to severe vitreous inflammation. Cytomegalovirus retinitis is quintessentially seen in the context of AIDS or severe immunosuppression. It still today remains an AIDS-defining infection but is also seen in patients who fail to respond, become intolerant of, or stop responding to highly active antiretroviral therapy (HAART). Traditionally, it was not associated with vitreous inflammation and therefore, detachments were rarely complicated by proliferative vitreoretinopathy. In the HAART era, the frequency of an inflammatory response, late detachments and PVR has increased with some series reporting up to 30% of CMV retinitis cases complicated by PVR.32 The incidence of RD is highest among newly diagnosed patients (within 45 days of diagnosing a CMV retinitis) at 4.9/100 eye years, while the overall rate among AIDS patients with CMV retinitis is 2.3/100 eye years.7 Additional risk factors for RD include large lesion size, anterior lesion location, associated retinal pathology (e.g., myopia) and older age.33,34 Local and systemic therapy might reduce slightly the risk of RD possibly due to a more rapid resolution of retinitis.35 Most detachments occur once the initial infection has subsided. The healed retinitis leaves a thin glial sheet above which an epiretinal membrane, possibly containing condensed vitreous, can be found (Fig. 112.5).36 Vitreoretinal gliosis is often present at the interface between normal and abnormal retina on optical coherence tomography (OCT) examination but is difficult to visualize clinically. It is possible that the gliosis contributes to the formation of retinal detachments in these patients.
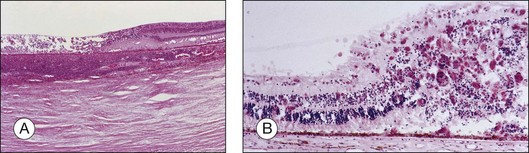
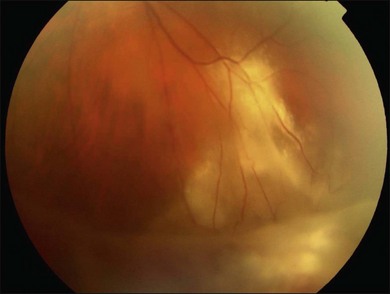
Acute retinal necrosis (ARN) is characterized by large, peripheral, often confluent zones of retinitis.37–39 It is pathologically characterized by full-thickness retinal necrosis and occlusive vasculitis (Fig. 112.4B).40 Vitreous involvement, often not present initially, develops within a few days and can obliterate the view of the posterior pole. A more aggressive form starting at the posterior pole called progressive outer retinal necrosis (PORN) affects also all retinal layers and represents an ophthalmic emergency since it may lead to complete blindness within hours if not treated aggressively.41–43 In both cases, detachments occur due to vitreous traction on the atrophic retina, sometimes following the occurrence of a PVD and when severe inflammation is present, they may occur even before resolution of the active retinitis. Retinal detachments in ARN are frequently complicated by PVR, which is often extensive at the vitreous base. Retinal breaks are usually multiple and located at the juncture between normal and gliotic retina. Vitreoretinal adhesions are common and the retinal detachment has both tractional and rhegmatogenous components (Fig. 112.6).
Clinical examination and findings
As stated above, RDs in uveitis can arise in a number of ways. Not infrequently, TRD, even schisis cavities, can be misdiagnosed as RRD (Fig. 112.7).16 As part of the initial work-up, it is important to establish the nature of the detachment, the etiology and severity of ocular inflammation or infection, and the status of the retina and vitreous. In particular, it is important to determine the relationship between the detached retina and the area of retinal inflammation or infection, the presence or absence of a PVD, and the presence of peripheral vitreous base fibrosis and foreshortening. These observations and work-up are often hampered by poor visualization resulting from posterior synechiae, cataract, vitreous haze or cellular infiltration. In infectious cases, particularly ARN, vitreous inflammation may increase shortly after initiating treatment to the point where it is impossible to visualize the fundus. Analogous to trauma cases, it is important in these cases to document, as early as possible, findings in the vitreous and retina.
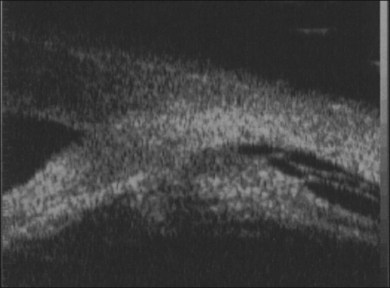
When visualization becomes inadequate, ancillary tests may be required. To evaluate the macular area through a small pupil, a nonmydriatic camera, SLO (scanning laser ophthalmoscopy), or OCT may be useful. Unfortunately, they do not give any information on the condition of the peripheral retina. B-scan ultrasonography (both 10 and 50 MHz) is probably the most useful tool for the evaluation of the peripheral fundus. Uveitis-related RD often has a highly reflective posterior hyaloid when detached (which can even mimic an RD).44 Occasionally, during active inflammation, fine echoes within the retrohyaloid space are visible, corresponding to inflammatory cells within the vitreous. If visualization of the vitreoretinal interface becomes difficult and therapy with intravitreal steroids is envisaged, triamcinolone will improve the ultrasound signal and facilitate visualization of posterior structures.45 The presence of a thickened choroid (≥2 mm thickness) and the asymmetry between the two eyes can help confirming the diagnosis of a choroidal inflammation in the involved eye.44 Choroidal detachments are not infrequent, and their anterior extent needs to be defined.
Ciliary body detachments may develop as a result of uveitis, causing or predisposing to hypotony. The UBM (ultrabiomicroscopy) is the preferred tool to visualize the ciliary body. Using a UBM, it is also possible to assess the peripheral vitreous, in particular if it has become adherent to the posterior iris surface causing posterior displacement of the iris plane (an indirect sign of vitreous base fibrosis and likely foreshortening of the retina) (Fig. 112.8).46,47
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


