Lymphoproliferative, Myeloproliferative, and Histiocytic Lesions of the Orbit
Omar M. Durrani
Rebecca Ford
David H. Verity
George Meligonis
Geoffrey E. Rose
Periocular Lymphoproliferative Diseases
Periocular lymphoid diseases range from benign hyperplasia to malignant lymphomas, the latter being solid tumors that arise from malignant transformation of leukocytes, particularly B lymphocytes (about 85%) or T lymphocytes, whereas malignancies without a distinct mass tend to be classified as leukemias. Lymphomas, of which about three quarters are non-Hodgkin’s B cell variants, comprise about 3% of systemic malignancies and generally arise in lymph nodes or primary lymphoid tissue, but may also occur in extranodal sites, such as the orbit, skin, oropharynx, gastrointestinal tract, or bone marrow. Lymphoma—almost exclusively low-grade non-Hodgkin’s tumors—is the most common orbital malignancy and accounts for 8% to 10% of all extranodal lymphomas. The World Health Organization (WHO) classification has replaced all previous classifications.1
Although orbital lymphatic vessels have been identified only in rhesus monkeys,2 tissue lymphocytes occur naturally within the conjunctival substantia propria, lacrimal gland, and lymphatics of the optic nerve dura; any lymphocytes outside these sites should be regarded as abnormal, whether part of an inflammatory or malignant process. Using monoclonal antibodies, the refinement of immunohistochemical techniques has vastly improved the understanding of lymphoproliferative diseases with better diagnosis, classification, prediction of prognosis, and characterization of phenotype for certain chromosomal anomalies.
Clinical Presentation and Diagnosis of Orbital Lymphocytic Lesions
Although an extranodal site in which lymphocytes are largely absent, orbital lymphoid lesions are the third most common cause of adulthood proptosis—after inflammation and cavernous hemangioma3—with an incidence of about 10% (7.5% of 764 orbital tumors; 11% of 820 cases).3,4 Low-grade orbital lymphomas usually present as a slowly progressive painless mass (Fig. 1A)) that, on imaging, molds to the globe and adjacent structures (Fig. 1B), but the much rarer higher grade lymphomas—such as diffuse large B cell lymphoma—may present as a painful, rapidly progressive mass with inflammatory signs (Fig. 2A) that need to be differentiated from orbital cellulitis. Very rarely, lymphoma will present with neuro-ophthalmic features from neighboring intracranial disease (Table 1).
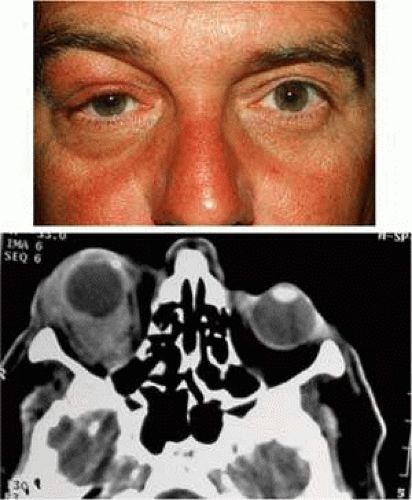 FIGURE 1. Low-grade orbital lymphoma–clinical signs. A. Slowly progressive painless proptosis. B. CT imaging identifying mass moulding the globe and adjacent structures. |
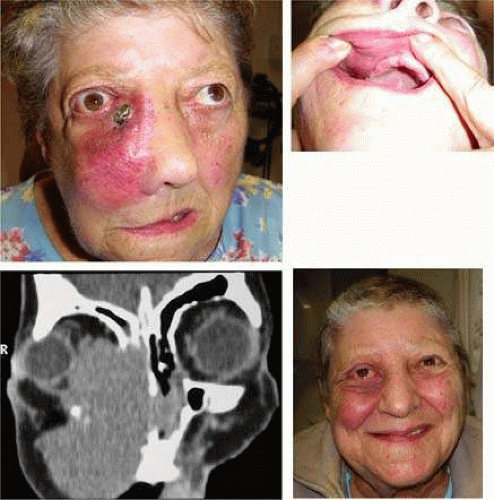 FIGURE 2. High-grade diffuse large B-cell orbital lymphoma–clinical signs. A. A 72-year-old patient presented with a 6-week history of rapidly progressive painful proptosis, diplopia, and cheek swelling. B. Invasion of the lesion into the cheek and upper alveolus. C. CT identifying a destructive lesion originating in the maxillary sinus and invading the orbit and ethmoid sinuses. A biopsy confirmed a diagnosis of diffuse large B-cell lymphoma. D. Remission following chemotherapy and radiotherapy. Despite an early response to treatment, the patient developed systemic metastases and died within 4 months. |
TABLE 1. Clinical Presentation of Orbital Lymphoproliferative, Leukemic, and Histiocytic Lesions | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||
Lymphomas generally display a fairly well-defined mass on CT, which is often centered on a major structure (such as the lacrimal gland) but extends with an irregular edge into neighboring tissues. Despite frequent loss of definition of normal orbital structures, these normal structures are almost never destroyed and many tissue boundaries, such as the globe or bone, are often respected macroscopically by the tumor invasion (Fig. 2B). The higher grade lymphomas often arise from neighboring structures, such as the paranasal sinuses (Fig. 2C), and secondarily invade the orbit—this wider involvement possibly being reflected in a worse prognosis.5 Bone molding, rather than invasion, may sometimes be seen with longstanding low-grade lymphomas, and diffuse calcification occurs only very rarely (Fig. 2D).
Systemic lymphoma will be present around the time of diagnosis in some patients presenting with orbital disease; the proportion is dependent on the histological type.5 All patients with orbital lymphoid lesions should undergo systemic evaluation with complete physical examination. Where appropriate, whole body imaging and bone marrow biopsy may be required. Bilateral orbital lymphoma has a significantly higher incidence of systemic involvement, although such systemic disease may be indolent and have only a small effect on life expectancy.6 Some low-grade lymphomas may lead to amyloid deposition in the orbit (Fig. 3), and others (such as the lymphoplasmacytoid variant) have been associated with the IgM secretion of Waldenström’s macroglobulinemia—this latter condition being associated with venous stasis and ischemic retinopathy secondary to a hyperviscosity syndrome.7
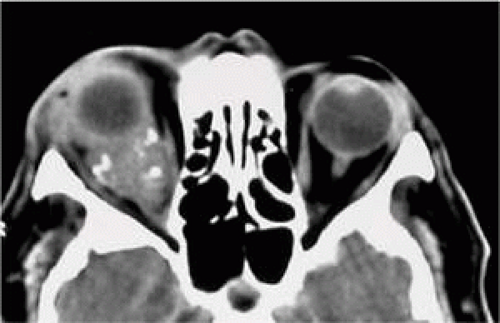 FIGURE 3. Amyloid deposition within the orbit in low-grade orbital lymphoma. |
Although fine-needle aspiration orbital biopsy may confirm the diagnosis of a lymphocytic lesion and the cellular aspirate further characterized by immunohistochemical studies, the best quality material for diagnostic pathology is acquired by open biopsy with minimal crushing of this rather friable tissue.
Types of Orbital Lymphocytic Lesions
About half of the primary orbital lymphoid lesions are reactive or atypical hyperplasia and half are malignant lymphoma,8 although there might be a continuous spectrum in terms of prognosis with systemic disease occurring in 15% to 25% of reactive hyperplasias, 40% of atypical hyperplasias, 20% of well-differentiated lymphomas, and up to 90% of poorly differentiated, high-grade lymphomas.2,5,8,9,10,11,12 It is probable that lymphoma emerges from a background of reactive lesions with gradual destruction of the reactive follicle by neoplastic lymphocytes, and this phenomenon might explain the paradoxical disease-related mortality rates of 6% for benign “reactive” hyperplasia, 19% for “atypical” hyperplasia, and 58% for malignant lymphoma.2 Overall, about a half of all patients with orbital lymphoid lesions—reactive, benign, or malignant—will have systemic involvement over a 4-year follow-up.2,8
Histologically, reactive (or “benign”) lymphoid hyperplasia (Figs. 4A-C) is the most innocuous of the lymphoid lesions and comprises a richly cellular tissue with sheets of mature polyclonal lymphocytes, scattered plasma cells, and histiocytes arranged in irregular follicles associated with hyperplastic capillaries lined by plump endothelial cells and little or no surrounding stroma.8 The germinal center consists of dendritic follicular cells, tingible body macrophages, and large lymphocytes with mitotic figures; this germinal center is surrounded by a mantle zone of 2 to 3 layers of small lymphocytes. In contrast, malignant lymphoma often shows little mitotic activity or endothelial proliferation and nodular or diffuse growth of a dense monoclonal population of uniform, immature lymphocytes of a small, medium, or large size. “Atypical” lymphoid hyperplasia, which may show atypical or aggressive clinical or histological features, shares some characteristics of both benign reactive hyperplasia and malignant lymphoma in that it lacks mitotic activity and endothelial proliferation, but does exhibit a follicular polymorphous response.8
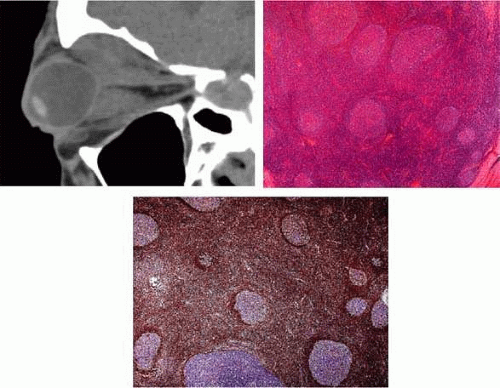 FIGURE 4. Reactive lymphoid hyperplasia. A. CT showing marked bilateral proptosis due to the presence of large infiltrative heterogenous density soft masses superiorly and inferiorly in the orbits. These lesions are largely intraconal, with spill over into the extra-conal compartments, engulfing both the inferior recti and superior recti and levator muscles complexes. The optic nerve sheath complexes are partially encompassed by the massive disease process. No definitional extension. B. In this case of reactive lymphoid hyperplasia, the follicles are round and well-demarcated by dark mantle zones. Tingible body macrophages are present in the germinal centers, which do not contain any atypical cells (H & E 20×). C: The germinal centers are negative on staining with Bcl-2 (20×). |
Approximately 85% to 90% of malignant orbital lymphomas are uniform throughout, of which about a half are well differentiated, and only 10% to 15% show distinct follicular structure.5 Patients with solely orbital lymphoma at presentation carry a significant risk of developing later systemic disease, this risk being related to the histological grade and continuing for up to 8 years after presentation.5
Orbital Lymphomas
Clinical features
Most orbital lymphomas are non-Hodgkin’s B cell variants, with a female preponderance (2:3 male:female ratio) and tend to present over the age of 50 years; presentation in childhood is extremely rare and, in this age group, leukemic infiltrates of the orbit would be much more common.
Orbital lymphoid lesions are usually located anterosuperiorly and present over several months with gradual painless proptosis of 5 mm or less, without conjunctival chemosis or injection (Fig. 1). Disturbance of orbital functions, such as visual function, ocular motility, globe displacement, or ptosis, may be mild or nonexistent, because (with the exception of orbital fat) lymphocytic lesions tend to mold around, rather than infiltrate, normal orbital structures. Involvement of the extraocular muscles has been described and most commonly involves the superior rectus–levator complex.13 Although there might be mild visual impairment in some cases,6 permanent loss is uncommon and bilateral blindness exceptional. Perhaps not unexpected with the mild lymphocytic population in normal lacrimal gland, this structure is involved in 26% of cases and typically the affected gland is palpable, mobile, and either firm or soft.6 Epibulbar lymphoid lesions typically display a pink coloration (“salmon patch”) and are freely mobile under the conjunctiva and over the bulbar surface (Fig. 5). Lymphadenopathy may be palpable and may indicate the presence of systemic disease. Knowles and Jakobiec report 64% of 117 lesions to be centered on the deep orbit, 28% subconjunctival, and 8% in the eyelid 6%.
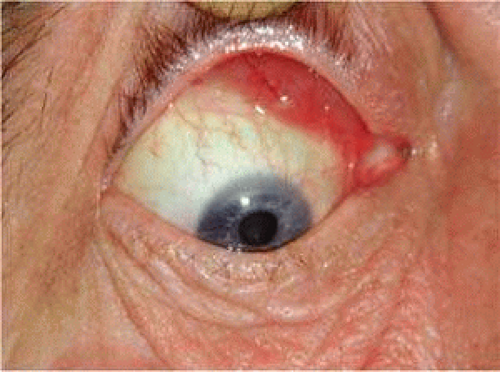 FIGURE 5. Pink mobile subconjunctival mass in the superonasal quadrant |
Classification of Lymphomas
As lymphoproliferative disorders comprise a diverse range of conditions that vary in presentation, prognosis, or treatment response, a practical, reproducible, and clinically useful classification is necessary to guide patient management. Although many classifications have been used in the past—the most commonly used of which was the Revised European-American Lymphoma (REAL) classification7 (Table 2) —a consensus grouping for hematological malignancies was published by the WHO in 2001,1 this WHO classification having worldwide acceptance and superceding all earlier classifications (Table 3). Tumors are stratified primarily according to lineage (into myeloid, lymphoid, histiocytic/dendritic cell, and mast cell groups); within each category, distinct diseases are defined according to a combination of morphological, immunophenotypic, and genetic features, and also by clinical syndromes.
TABLE 2. The Revised European American Lymphoma Classification (REAL) | |
|---|---|
|
TABLE 3. World Health Organization (WHO) Lymphoma Classification with ICD-0-3 Codes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
1.3.3 Etiopathology of Lymphomas
Multiple theories exist regarding the etiology and pathogenesis of lymphoma. Although infectious agents have been postulated as a risk factor for lymphoma, no specific pathogens have been identified. A higher risk of non-Hodgkin’s lymphoma (NHL) has been noted in patients with rheumatoid arthritis or in those on aspirin or other nonsteroidal anti-inflammatory drugs (NSAIDs) and, conversely, 131 patients with NHL showed a positive association with NSAID usage.14 The increased risk of NHL with rheumatoid arthritis might result from suppression of cellular immunity by inhibition of prostaglandin E2 synthesis, or possibly because of the underlying autoimmune disease, or the long-term immunosuppressives used for treatment. Several autoimmune diseases, such as Sjögren’s syndrome, Hashimoto’s thyroiditis, and Crohn’s disease, predispose a patient to systemic lymphoma and prior thyroid eye disease appears to predispose a patient to orbital lymphoma about 15 or more years later.15,16
Improving techniques in molecular biology have improved our understanding of the morphology and functioning of lymphoma cells, with development of immunopathologic techniques useful in lymphoma diagnosis and classification, and also a greater appreciation of the cytogenetic changes underlying lymphomagenesis.17,18
Immunopathology
Lymphomas arise from clonal proliferation of lymphocytes at various stages of differentiation from precursor T and B cells to mature T cells and plasma cells: B cells arise from the bone marrow and produce immunoglobulins in a humoral response, whereas T cells are thymus-derived and respond in a cell-mediated reaction via cells that are antigenically marked or that produce lymphokines. Lymphocytes in circulating blood and lymph nodes normally have a ratio of about two thirds T cells to one third B cells, natural killer (NK) cells, and null cells. The normal lymph node comprises follicles, each organized into germinal centers, mantle zones, and the surrounding cortex (Fig. 6). The germinal center is composed of a framework of dendritic histiocytes, follicular dendritic cells, centroblasts, centrocytes, immunoblasts, small lymphocytes, and tingible body macrophages, whereas the surrounding cortex and paracortex contain mainly B and T cells, respectively.
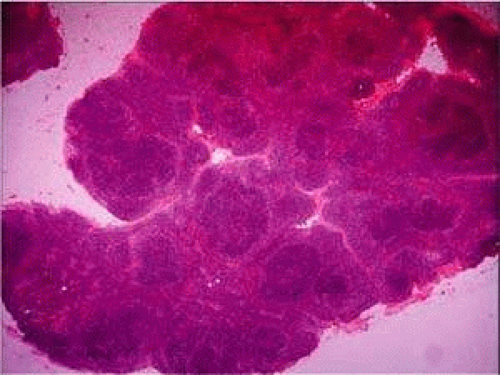 FIGURE 6. Lymph node. Lymph node showing several reactive germinal centers predominantly in a cortical location. The node has a thin fibrous capsule peripherally and is surrounded by adipose tissue. A small segment of an adjacent lymph node is seen the in the top left corner. (Courtesy Dr. Anna Childerhouse.) |
As lymphomas arise from monoclonal proliferations, histological demonstration of a single-cell type aids diagnosis and classification and, hence, the planning of treatment and determination of prognosis. Histological identification of lymphomatous B cells was originally achieved with heterosera against immunoglobulins, these sera being counterstained with immunoperoxidase stain or labelled with immunofluorescent markers.19 Plasma cells could be similarly identified by their cytoplasmic immunoglobulins. T cells were first identified cytologically by rosette formation when combined with sheep erythrocytes but, in addition, have been noted to uniquely contain cytoplasmic α-naphthyl esterase, for which the cells can be stained.20,21 More specific and sensitive immunohistochemical and cytogenetic techniques are now available, allowing further immunophenotyping of the cellular populations within the tumor biopsy.18
B-cell clonality can be indicated by quantitative histochemical staining for the kappa and lambda immunoglobulin light chains: the kappa–lambda ratio in benign lesions is 2:1, whereas the ratio is typically 10:1 or greater in malignant lesions.21 Orbital lymphomas are almost exclusively B-cell proliferations and contain a low proportion (10% to 20%) of T cells, whereas over a half of all lymphocytes within benign or atypical reactive lesions are T cells; in contrast, T-cell lymphomas contain around 90% T cells.21 The normal T-cell “helper:suppressor” ratio is 2:1, and this is maintained in malignant B-cell lymphomas, but typically increases to 5:1 in reactive lymphoid lesions.21 Reactive processes involve polyclonal or oligoclonal populations, in which each of the many clonal populations expresses a distinct gene rearrangement, whereas a malignant lymphoma arises from a monoclonal clone population—thereby expressing only a single immunoglobulin genomic rearrangement—these gene rearrangements being characterized by polymerase chain reaction (PCR) methods.
Cell surface markers have been identified for various cell types—initially the immunoglobulins and interleukins and, later, the cell surface antigen receptors—these being termed clusters of differentiation (CD); the CD markers are used to characterize the degree of cellular differentiation.22,23 There are currently well over a hundred characterized cell surface markers, providing valuable help in the characterization of Hodgkin’s lymphoma and the subsets of non-Hodgkin’s B- and T-cell lymphomas (Table 4).24 Some CD markers are typical of B cells, such as CD10, CD19, CD20, and CD22, whereas others (such as CD2, CD4, CD7, and CD8) are expressed by T cells.
TABLE 4. List of Lymphoid Lesions and Their Typical Immunophenotype Cell Markers | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
Cytogenetics of Lymphoma
Cytogenetic analysis now plays a significant role in the diagnosis of some lymphomas, as many such tumors may result from a failure of the regulation of somatic mutations either as a loss of suppressor gene activity or as an upregulation of a proto-oncogene that promotes mitosis with a differentiation of naive lymphocytes to memory cells and plasma cells. Changes in genetic control probably also explain the increased risk of developing non-Hodgkin’s lymphoma in immunocompromised patients with congenital immunodeficiencies, autoimmune diseases, on cytotoxic agents, or with acquired immunodeficiency syndrome (AIDS). Lymphomas in AIDS patients tend to be clinically aggressive—with involvement of bone marrow, bowel, and the central nervous system—and are associated with a poor prognosis.16,25,26,27,28,29
Immunoglobulin gene expression has been studied in lymphoma, and conventional cytogenetic analysis provides information about chromosomal rearrangements within tumor cells. Gene rearrangements can be detected with restriction endonuclease cleavage and Southern blot techniques,30,31,32,33,34,35,36,37 the rearrangement appearing as a distinct and unique band with monoclonal lesions, whereas a polyclonal lesion, having immunoglobulins from many different lymphocyte populations, demonstrates only the control band.30,31,32,33,36,37 The technical difficulties with conventional techniques have, however, led to increased use of PCR or fluorescence in-situ hybridization (FISH) analysis to identify specific genetic abnormalities; multicolor interphase FISH assays allow detection of several diagnostic changes in a single cell, and simultaneous fluorescence immunophenotyping (FICTION technique) facilitates a correlation of cellular phenotype and genotype.18
Many cytogenetic abnormalities, mostly chromosomal translocations, have been shown to be characteristic for particular lymphomas, and these are particularly associated with upregulation of proto-oncogenes and transcription factors or with downregulation of apoptosis. Multiple chromosomal translocations are an integral part of normal B-cell differentiation, and this is thought to explain the high frequency of such genetic abnormalities within malignant lymphomas (Table 5). Examples of such mutations include that of the c-myc oncogene (8q24) in Burkitt’s lymphoma, the bcl-1 gene (11q13) in mantle cell lymphoma, the bcl-2 translocation (14;18)(q32;q21) in 70% to 90% follicular lymphomas and 10% to 35% of nodal large-cell lymphomas, and the bcl-6 gene (3q27) in diffuse large B-cell lymphoma.37,38,39,40,41,42,43,44
TABLE 5. Lymphoid Lesions and Their Characteristic Gene Rearrangements and Mutations | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
The specific genes overexpressed or underexpressed as a result of some mutations or translocations have begun to be elucidated: Half of lymphoplasmacytic lymphomas, often with macroglobulinemia, are associated with a (9;14)(p13;q32) translocation, this translocation juxtaposing the immunoglobulin heavy chain (IgH) gene (on chromosome 14q32) and the PAX-5 gene (9p13). PAX-5 encodes the cell cycle regulation protein B-cell specific activation protein (BSAP)—a control protein involved throughout B-cell proliferation and differentiation with the exception of plasma cells (Table 4).43 The PAX-5 gene belongs to a group of PAX genes, which have been identified as regulators of cell cycle and differentiation,43,45 and BSAP factor may enhance B-cell lymphopoiesis by an unknown mechanism. The protein product of the bcl-2 gene reduces cellular apoptosis, and a (14:18)(q32;q21) translocation with increased bcl-2 protein production confers a survival advantage to the centrocytes in follicular lymphoma.46 The c-myc gene is known to code for a proto-oncogene which, when upregulated, allows for unmitigated cell division.7,47 The bcl-1 gene mutation allows upregulation of the PRAD-1 gene that encodes a cell cycle regulator, cyclin D1.47 Anaplastic large cell lymphoma (ALCL) is typically associated with the t(2,5)(p23;q35) translocation, which includes fusion of the NPM (nucleophosmin) gene on 5q35 to a novel anaplastic lymphoma kinase (ALK) gene on 2p23, which results in phosphorylation of intracellular targets and triggers malignant transformation.48 The ALK gene is not expressed on normal T cells and has both diagnostic and prognostic importance.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


