Chapter 44 Inherited retinal disorders
Introduction
The inherited retinal disorders are a clinically and genetically heterogeneous group of conditions: many become symptomatic in childhood, occurring as an isolated abnormality in an otherwise healthy child. Some are associated with systemic abnormalities (see Chapter 45). Most of the genes causing the major childhood retinal dystrophies have been identified: the genotype–phenotype relationship is complex. There is considerable genetic heterogeneity for individual clinical disorders, and mutations in a single gene may give rise to several different phenotypes. Despite this, these disorders can be usefully divided clinically according to whether they:
Stationary retinal dysfunction syndromes
Stationary night-blindness (rod dysfunction syndromes)
Congenital stationary night-blindness
Clinical findings
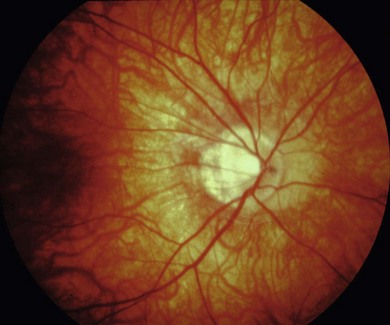
The visual acuity is usually normal or mildly reduced in the AD form, whereas mild to moderate central visual loss is common in the AR and XL subtypes. Other features of XL and AR CSNB include moderate to high myopia, nystagmus, strabismus, and paradoxical pupil responses. Fundus examination is usually normal but some patients have myopic fundi and pale or tilted optic disks (Fig. 44.1). Patients with AD CSNB usually present with symptomatic night-blindness, but in XL and AR CSNB, patients usually present in infancy with nystagmus, strabismus, and reduced vision. Nystagmus is not invariable and some patients are not diagnosed until late childhood or adulthood. The diagnosis is easily missed without electroretinography (ERG). XL and AR CSNB may be further subdivided into complete and incomplete forms. This differentiation was originally proposed in XL disease using electrophysiologic and psychophysical criteria and was subsequently shown to reflect genetically distinct disorders.
Electrophysiology
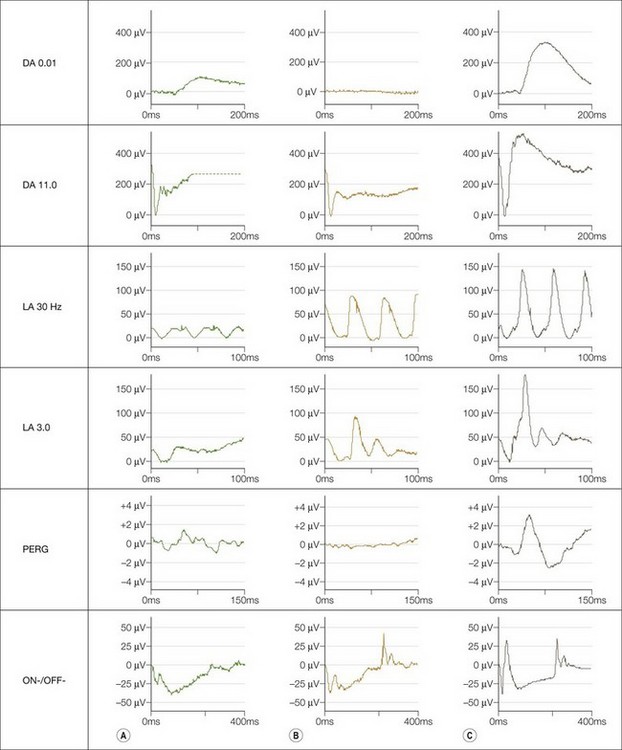
International Society for Clinical Electrophysiology of Vision (ISCEV) standard ERGs should be performed. This may not be possible in infants in whom a modified protocol is used (see Chapter 8). Four main responses are defined: a rod-specific ERG and a bright flash response performed under scotopic conditions, and two measures of cone function, a 30 Hz flicker ERG and a single flash photopic ERG. Both complete and incomplete CSNB show a “negative ERG”: the photoreceptor-derived a-wave in the bright flash response is normal, but there is selective reduction in the inner nuclear derived b-wave, such that it is smaller than the a-wave, indicating predominantly inner retinal dysfunction. In complete CSNB there is no detectable rod-specific ERG and a profoundly negative bright flash response. Cone ERGs show subtle abnormalities reflecting ON bipolar cell dysfunction (Fig. 44.2). There is a detectable rod-specific ERG in incomplete CSNB, and a profoundly negative bright flash response. Cone ERGs are much more abnormal than in complete CSNB, reflecting involvement of both ON- and OFF bipolar pathways. They show the characteristic triphasic appearance in the flicker response (see Fig. 44.2).
Molecular genetics and pathogenesis
X-linked CSNB
Several genotype–phenotype studies have been performed in individuals with either CACNA1F or NYX mutations. There is considerable inter- and intra-familial phenotypic variability associated with CACNA1F mutations, even with an identical sequence variant,1 suggesting that other genetic or environmental factors modify the phenotype. Although most patients with XL CSNB have non-progressive disease, Nakamura et al. reported two brothers with a CACNA1F mutation and progressive decline in vision with eventually undetectable rod and cone ERGs.2 We have also infrequently observed slow progression in patients with XL CSNB. Patients with complete CSNB (NYX mutations) are invariably myopic and have more pronounced night-blindness.3
Autosomal recessive CSNB
Sequence variants have been identified in SLC24A1, in patients with AR CSNB without a negative ERG; in the standard scotopic bright flash response, both the a- and b-waves are equally reduced.4 SLC24A1 is a member of the solute carrier protein superfamily located in inner segments, outer and inner nuclear layers, and ganglion cells.
Åland Island eye disease
Åland Island eye disease (AIED) is an X-linked recessive disorder similar to incomplete CSNB, characterized by reduced visual acuity, nystagmus, nyctalopia, mild red–green dyschromatopsia, and myopia. Affected males may show iris translucency, foveal hypoplasia, and decreased fundus pigmentation. The clinical appearance may resemble X-linked ocular albinism (XLOA) but in XLOA color vision is usually normal and patients with AIED do not show the typical optic nerve fiber misrouting seen in albinism.5
Oguchi’s disease
Clinical findings
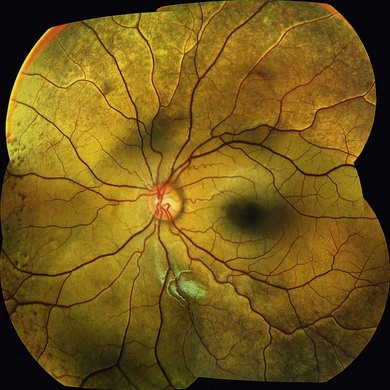
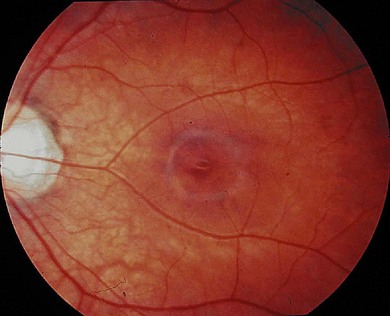
Oguchi’s disease is a rare autosomal recessive stationary night-blindness with a greyish or green-yellow discoloration of the fundus at the posterior pole or extending beyond the arcades, which reverts to normal on prolonged dark adaptation (Mizuo phenomenon) (Fig. 44.3). Exposure to light then usually leads to the gradual reappearance of the abnormal discoloration in 10–20 minutes.
Electrophysiology and psychophysics
There are two types of Oguchi’s disease depending on the dark adaptation findings:
Type 1: rod adaptation is markedly slowed; full recovery of sensitivity takes several hours and the absolute threshold is normal or only minimally elevated.
Type 2: there is no recognizable rod adaptation; the abnormal retinal appearance is less marked and the Mizuo phenomenon may be absent.
Molecular genetics and pathogenesis
Null mutations in GRK1, encoding a second component of the rod phototransduction pathway, rhodopsin kinase (RK), have also been identified in Oguchi’s disease. The key function, of both RK and arrestin, in the normal deactivation and recovery of the photoreceptor after exposure to light, explains the delayed recovery in Oguchi’s disease. The consequences of these reported RK mutations have been assessed by expression studies in COS7 cells of wild-type and human mutant RK. The markedly reduced mutant RK activity supported their pathogenicity.6
Evidence from knockout mice models suggests that patients with SAG or GRK1 mutations may be more susceptible to light-induced retinal damage; it may therefore be advisable for patients to wear tinted spectacles.7,8
Fundus albipunctatus
Clinical findings
The deposits are discrete dull white lesions at the level of the retinal pigment epithelium (RPE). They are most numerous in the mid-periphery and are usually absent at the macula; the optic discs and retinal vessels are normal. Fluorescein angiography shows multiple areas of hyperfluorescence, which may not directly correlate to the deposits seen clinically. In some patients there is good correlation between the white dots and increased autofluorescence on fundus autofluorescence imaging, but not in others. The differential diagnosis is from other causes of flecked retina (see Chapter 48).
Electrophysiology and psychophysics
Dark adaptation is severely delayed in fundus albipunctatus (FA), reflecting abnormal regeneration of rhodopsin. The rod-cone break is delayed and full rod adaptation may take many hours. Rod ERGs are markedly abnormal, with the rod-specific ERG (DA 0.01) being undetectable under standard conditions, but becoming normal following prolonged dark adaptation (Fig. 44.4). The dark-adapted bright flash ERG (DA 11.0), which after standard dark adaptation arises in dark-adapted cones, can have a low b : a ratio; a red flash stimulus under dark adaptation shows a normal cone component but an undetectable rod component and prevents confusion with a form of CSNB associated with a negative ERG. To confirm the diagnosis of FA it is necessary to exceed the ISCEV ERG standard recommendations for dark adaptation considerably. Most but not all patients with RDH5 mutations show full recovery of rod function with extended dark adaptation. This contrasts with the findings in retinitis punctata albescens (see below), related to mutation in RLBP1, and usually allows the distinction between the two disorders.
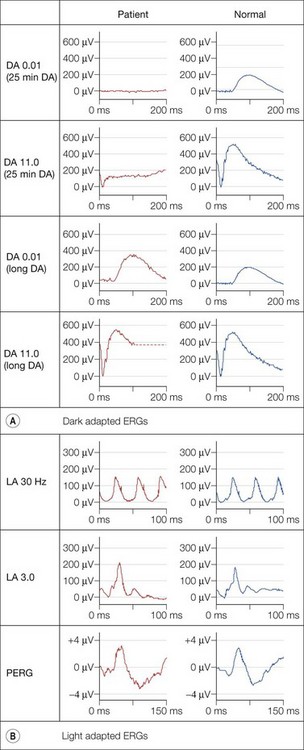
There are two forms of FA, one in which cone ERGs are normal, and a rarer form described as fundus albipunctatus with cone dystrophy and negative ERG.9
Molecular genetics and pathogenesis
Mutations in three genes (RDH5, RLBP1, and RPE65), encoding components of the visual cycle, have been identified to date. The gene RDH5 encodes 11-cis-retinol dehydrogenase, a component of the visual cycle. Recombinant mutant 11-cis retinol dehydrogenases have reduced activity compared with recombinant enzyme with wild-type sequence.10 The phenotype of patients with RDH5 mutations is variable, including normal or reduced cone responses.11 The function of the protein product of RDH5 is consistent with the delay in the regeneration of photopigments characteristic of the disorder.
Sequence variants in RPE65 have also been identified in patients with a retinal appearance similar to FA.12 Reduced fundus autofluorescence has been observed in patients with RPE65 and RDH5 mutations,11,12 supporting the belief that disruption of retinoid recycling in the RPE is essential for the development of FA.
Stationary cone disorders (cone dysfunction syndromes)
The cone dysfunction syndromes include congenital color vision disorders where there is normal visual acuity but defective color vision, and the various forms of cone dysfunction associated with reduced central vision and often nystagmus and photophobia (Table 44.1).13
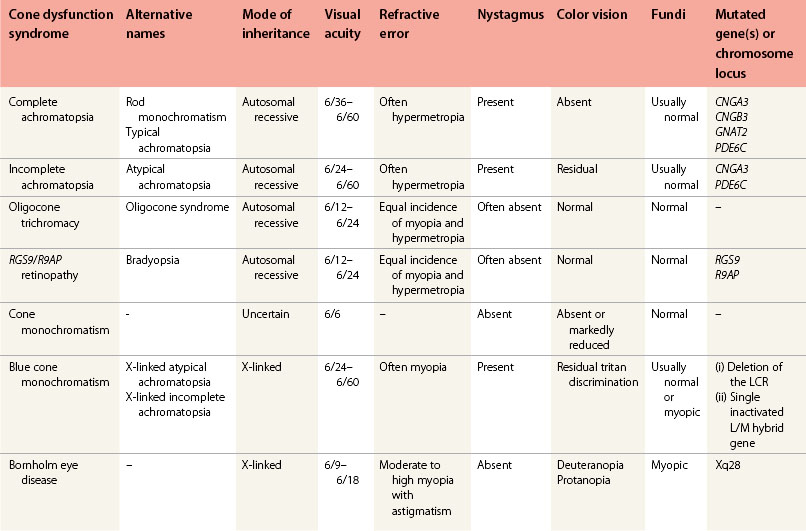
Disorders of color vision with normal visual acuity
This group of disorders, their clinical characteristics, and the molecular pathology are reviewed elsewhere.14
Achromatopsia
Achromatopsia is genetically heterogeneous and characterized by an absence of functioning cones in the retina.13 It may occur in complete (typical) and incomplete (atypical) forms.
Complete achromatopsia (rod monochromatism)
Clinical and histopathologic findings
The visual acuity is usually 6/60 and there is complete color blindness. Peripheral visual fields are normal but a small central scotoma can often be detected. Achromatopsia is generally a stationary disorder but slow deterioration and macular atrophy may occur.15,16
Histopathologic studies have demonstrated cone-like structures in the retina,17,18 which have also been observed in vivo using adaptive optics imaging, albeit to a variable degree.16 This is promising with regard to gene replacement therapy.
Electrophysiology and psychophysics
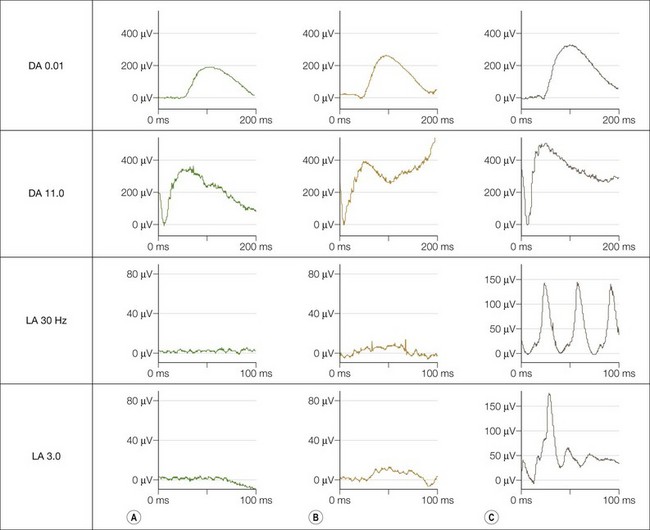
The dark adaptation curve is monophasic with no evidence of a cone contribution and spectral sensitivity studies show that rods mediate threshold under both photopic and scotopic conditions; there is no evidence of a Purkinje shift. Electroretinography (Fig. 44.5) shows normal rod-derived ERGs but no detectable cone-derived responses.
Molecular genetics and pathogenesis
More than 60 disease-causing mutations in CNGA3 have been identified in patients with achromatopsia; with four mutations (Arg277Cys, Arg283Trp, Arg436Trp, and Phe547Leu) accounting for approximately 40% of all mutant CNGA3 alleles.19 By comparison, far fewer mutations have been identified in CNGB3; with the 1 base-pair frameshift deletion, 1148delC (Thr383fs), accounting for up to 80% of CNGB3 mutant disease chromosomes.20,21 The majority of CNGA3 mutations identified to date are missense mutations, indicating that there is little tolerance for substitutions with respect to functional and structural integrity of the channel polypeptide. In contrast, the majority of CNGB3 alterations are nonsense mutations.
About 70% of achromatopsia results from mutations of CNGA3 and CNGB3, with GNAT2 and PDE6C each accounting for less than 1%;19,20,21 further causative genes remain to be discovered.
Incomplete achromatopsia
Molecular genetics and pathogenesis
As in the complete form, mutations in CNGA3 have been identified in individuals with incomplete achromatopsia.19 The mutations identified are all missense mutations, located throughout the channel polypeptide including the transmembrane domains, ion pore, and cGMP-binding region. Only three of these, Arg427Cys, Arg563His, and Thr565Met, are exclusively found in patients with incomplete achromatopsia.19 Therefore, in the majority of cases of incomplete achromatopsia, other factors may influence the phenotype such as modifier genes or environmental factors. The missense variants identified in incomplete achromatopsia must be compatible with residual channel function since the phenotype is milder than in complete achromatopsia. Mutations in PDE6C have also been identified in incomplete achromatopsia.22
S-cone monochromatism (blue cone monochromatism)
Clinical findings
Blue cone monochromatism (BCM) is an X-linked recessive disorder, affecting approximately 1 in 100 000 individuals, in which affected males have normal rod and blue (S) cone function but lack red (L) and green (M) cone function. The clinical features are similar to complete achromatopsia but less severe. Affected infants have photophobia and develop small amplitude rapid nystagmus in early infancy. They are usually myopic, in contrast to achromatopsia (Fig. 44.6). The nystagmus reduces with time.
BCM is generally accepted to be a stationary disorder but deterioration and the development of macular atrophy may occur.23,24,25,26
Electrophysiology and psychophysics
Achromatopsia and BCM may be differentiated by the mode of inheritance, findings on psychophysical testing and electrophysiology. There is some preservation of the single flash photopic ERG (LA 3.0) in BCM (Fig. 44.5), and specialised spectral ERG techniques to measure S-cone ERGs can also be used (see Fig. 44.5). Female carriers of X-linked BCM may have abnormal cone ERGs and mild anomalies of color vision.
Molecular genetics and pathogenesis
Mutation analyses have established the molecular basis for BCM.24 The mutations in the L- and M-opsin gene array cause BCM to fall into two main classes:
1. A normal L- and M- pigment gene array is inactivated by a deletion in the locus control region (LCR), located upstream of the L-pigment gene. A deletion here abolishes transcription of all genes in the pigment gene array and therefore inactivates both L- and M-cones.
2. The LCR is preserved but changes within the L- and M-pigment gene array lead to loss of functional pigment production. The most common genotype in this class consists of a single inactivated L/M hybrid gene. The first step in this second mechanism is unequal crossing over reducing the number of genes in the array to one, followed in the second step by a mutation that inactivates the remaining gene. The most frequent inactivating mutation that has been described is a thymine-to-cytosine transition at nucleotide 648, which results in a cysteine-to-arginine substitution at codon 203 (Cys203Arg), a mutation known to disrupt the folding of cone opsin molecules.
The data suggest that 40% of BCM genotypes are a result of a one-step mutational pathway that leads to deletion of the LCR. The remaining 60% of BCM genotypes comprise a heterogeneous group of multi-step pathways. Studies have not detected the genetic alteration that would explain the BCM phenotype in all assessed individuals.24 In one study the structure of the opsin array did not reveal the genetic mechanism for the disorder in 9 of 35 affected patients24 which may suggest genetic heterogeneity yet to be identified in BCM.
Oligocone trichromacy
Oligocone trichromacy is characterized by reduced visual acuity (6/12 to 6/24), mild photophobia, normal fundi, reduced cone ERGs with normal rod responses, and normal color vision.13 Patients have been described both with and without nystagmus. These patients may have a reduced number of normal functioning cones (oligocone) with preservation of the three cone types in the normal proportions, thereby permitting trichromacy. High-resolution quantitative retinal imaging supports this.27
Oligocone trichromacy is inherited as an autosomal recessive trait; the molecular genetic basis is unknown. Patients with either RGS9/R9AP mutations (“bradyopsia”; see below) or oligocone trichromacy have very similar clinical phenotypes, characterized by stationary cone dysfunction with normal color vision.28 The distinctive electrophysiologic features associated with RGS9 and R9AP mutations enable directed genetic screening.
RGS9/R9AP retinopathy (bradyopsia)
Clinical findings
Bradyopsia is a stationary cone dysfunction syndrome with onset in early childhood; it is characterized by mild photophobia, markedly delayed dark and light adaptation, moderately reduced visual acuity, normal color vision, and normal fundi. Some patients report difficulty seeing moving objects. It is not possible clinically to distinguish bradyopsia from oligocone trichromacy; however there are pathognomonic findings on extended electrophysiologic assessment.28
Bornholm eye disease
Clinical findings
Bornholm eye disease is an X-linked disorder with moderate to high myopia and astigmatism, impaired visual acuity, moderate optic nerve hypoplasia, thinning of the RPE in the posterior pole with visible choroidal vasculature, and abnormal cone ERGs.29 Affected members in this single family were all deuteranopes, with a stationary natural history; however a similar phenotype has also been published in association with protanopia.30,31 This disorder is therefore best characterized as an X-linked cone dysfunction syndrome with myopia and dichromacy.
Progressive retinal dystrophies
Rod–cone dystrophies
The rod–cone dystrophies (retinitis pigmentosa (RP)) are a clinically and genetically heterogeneous group of disorders with progressive loss of rod and later cone photoreceptor function leading to severe visual impairment. RP usually occurs as an isolated retinal abnormality, but it may also be seen with systemic abnormalities (see Chapter 45).
Leber’s congenital amaurosis
Clinical findings
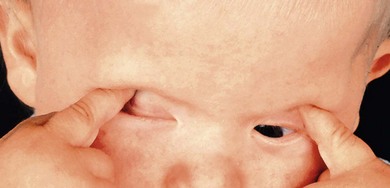
Severe visual impairment is from birth or the first few months of life with roving eye movements or nystagmus and poor pupillary light responses. Eye-poking, the “oculodigital” sign, is common (Fig. 44.7). Fundus examination may be normal but a variety of abnormal fundus appearances may be present such as disk pallor, vessel attenuation, or mild peripheral pigmentary retinopathy. There may also be disk drusen, optic disk edema or pseudopapilledema (Fig. 44.8), a flecked retina, maculopathy, or nummular pigmentation. Affected infants often have high hyperopia, or less commonly high myopia, suggesting some interference with emmetropization.
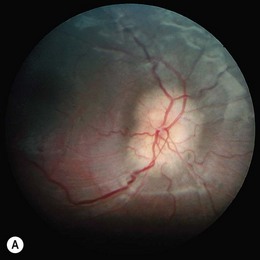
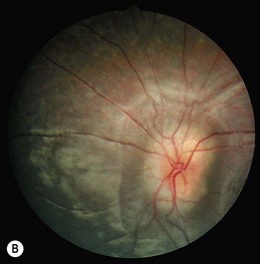
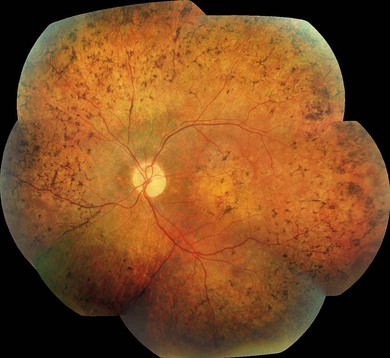
Following the discovery of many disease-causing genes (below) it has sometimes been possible to identify characteristic associated phenotypes (Figs 44.9 to 44.12): RDH12-associated disease is characterized by bone spicule pigmentation and maculopathy (Fig. 44.9) and CRB1-associated disease has nummular pigmentation, maculopathy, relative preservation of para-arteriolar RPE, with retinal thickening and loss of lamination on optical coherence tomography (Figs 44.10 and 44.11).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


