Chapter 46 Inherited macular dystrophies
Introduction
The inherited macular dystrophies are characterized by bilateral central visual loss and symmetrical macular abnormalities. Most present in the first two decades of life with a wide range of clinical, electrophysiological, psychophysical, and histological findings. The molecular basis of inherited macular disease is now well understood, providing insights into the pathogenesis (Table 46.1).
We review pediatric macular dystrophies and not those that present later, such as Sorsby’s fundus dystrophy, dominant drusen, and adult vitelliform macular dystrophy. Systemic disorders with macular dystrophy will be discussed in Chapters 45 and 62.
Autosomal recessive inheritance
Stargardt’s disease and fundus flavimaculatus
Clinical and histological findings
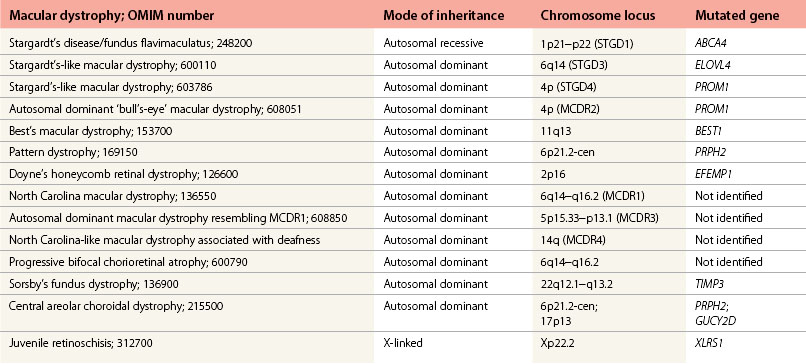
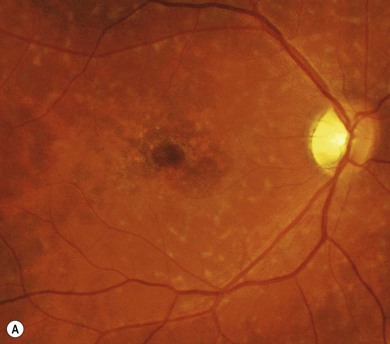
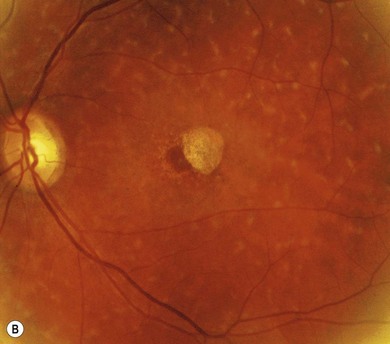
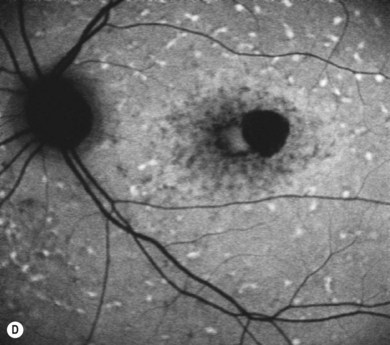
Stargardt’s macular dystrophy (STGD) is the most common inherited macular dystrophy with a prevalence of 1 in 10 000; it is inherited as an autosomal recessive trait. Most cases present with central visual loss in early teens. There is typically macular atrophy with yellow-white flecks at the level of the retinal pigment epithelium (RPE) at the posterior pole (Fig. 46.1). The flecks may be “fish-shaped” (pisciform), round, oval, or semilunar. The oval area of macular atrophy may, in the early stages, have a “beaten bronze” appearance (see Fig. 46.1). However, there may be no evidence of flecks at presentation, with the only abnormality being macular atrophy; but usually flecks develop over time. Fundus flavimaculatus (FFM) is when the retinal flecks occur without macular atrophy. STGD and FFM are caused by mutations in the same gene; both patterns may be seen in the same family. Most patients who present with FFM develop macular atrophy.
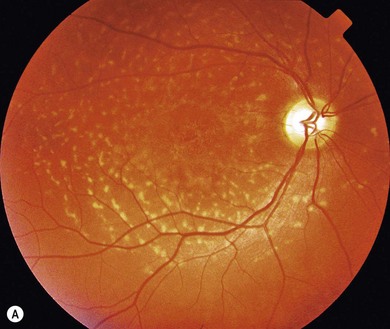
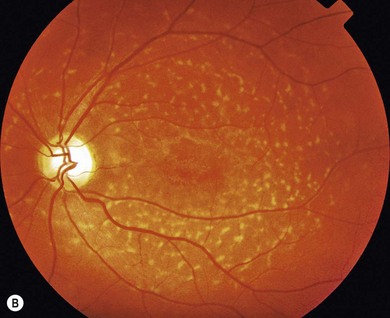
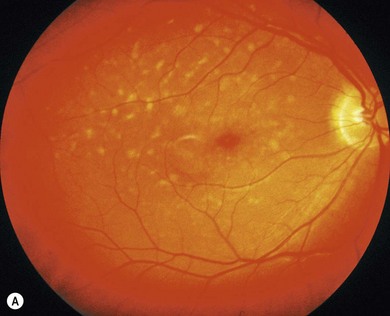
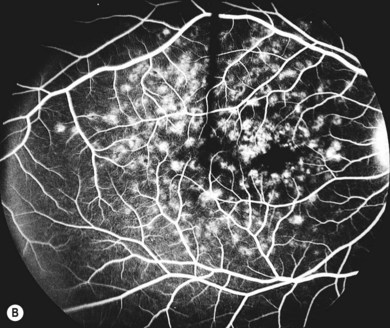
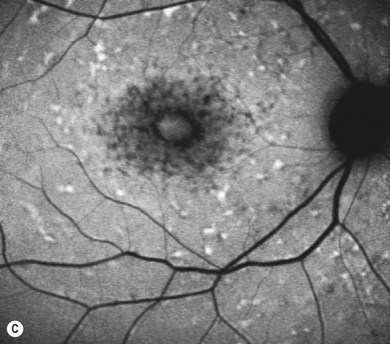
In both STGD and FFM, fluorescein angiography classically reveals a dark or masked choroid (Fig. 46.2) in the early phase. This is due to excess lipofuscin accumulation in the RPE, which obscures fluorescence from choroidal capillaries. The retinal flecks appear hypofluorescent on FFA early in their evolution, but later they appear hyperfluorescent due to RPE atrophy. Autofluorescence (AF) imaging, using the intrinsic fluorescence from lipofuscin in the RPE, has superseded FFA in confirming the diagnosis. The abnormal accumulation of lipofuscin, the presence of active and resorbed flecks, and RPE atrophy are characteristic on fundus AF imaging.1 (Fig. 46.3). In children with normal fundoscopy and visual loss from macular dysfunction, FFA is still helpful; a subtle window defect in the central macula or a dark choroid helps confirm the diagnosis.
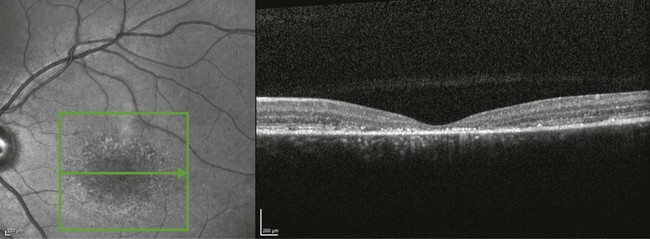
Optical coherence tomography (OCT) often reveals loss, or marked disruption, of central outer retinal macular architecture with relative preservation of peripheral macular structure (Fig. 46.4).
Electrophysiology
Electrophysiological abnormalities in STGD are variable. An abnormal electro-oculogram (EOG), suggestive of generalized RPE dysfunction, is common. The pattern electroretinogram (PERG) and focal ERG are usually abolished or markedly reduced, suggesting macular dysfunction. The full-field ERG may be normal at diagnosis (group 1) or suggest widespread retinal dysfunction (group 2 or 3):1
Group 1: severe pattern ERG abnormality with normal full-field ERG.
Group 2: additional generalized cone dysfunction.
Molecular genetics and pathogenesis
Mutations in the gene ABCA4 underlie STGD/FFM, and have also been implicated in retinitis pigmentosa (RP) and cone−rod dystrophy. ABCA4 encodes a transmembrane rim protein in the outer segment discs of rod and cone photoreceptors involved in transport of retinoids from photoreceptor to RPE. Failure of this transport results in deposition of a lipofuscin fluorophore, A2E (N-retinylidene-N-retinylethanolamine), in the RPE,2 which is deleterious to the RPE and leads to secondary photoreceptor degeneration.
Correlation between the type and combination of ABCA4 mutations with the severity of the phenotype is often possible.3 For example, biallelic null mutations usually lead to a cone−rod dystrophy phenotype rather than STGD. Variable retinal phenotypes within families may be explained by different combinations of ABCA4 mutations segregating within a single family; it is likely that other modifier genes or environmental factors may also influence intra-familial variability.
Accumulation of lipofuscin-related products in the RPE, such as A2E, occurs in STGD and in ABCA4 knockout mice (abca4−/−), resulting in free radical generation, release of pro-apoptotic mitochondrial proteins, and lysosomal dysfunction.2 This leads to RPE dysfunction and cell death and subsequent photoreceptor cell loss.
Future therapies
New therapeutic interventions for STGD include drugs that target the ATP dependent transport mechanism thereby augmenting ABCA4-related retinoid transport, or slow the visual cycle, reducing the production of A2E. Directly inhibiting the toxic effects of A2E may prove more effective. Pharmacological agents aimed at these three targets have been developed and are likely to progress to a human clinical trial in the near future.2,4 Such agents may also be helpful in other macular degenerations associated with lipofuscin accumulation, such as Best’s disease.
Other interventions include gene supplementation (http://www.clinicaltrials.gov), and cell-based or stem cell-based strategies aimed at providing supportive growth factors or RPE/photoreceptors for transplantation, respectively. It is likely that cell/stem cell-based clinical trials will be undertaken soon.
Autosomal dominant inheritance
Autosomal dominant Stargardt’s-like macular dystrophy
Electrophysiology
Individuals with AD-STGD-like dystrophy often have no significant EOG or ERG abnormalities.
Autosomal dominant “bull’s-eye” macular dystrophy (MCDR2)
Clinical and histological findings
Onset is from the end of the first to the sixth decade, most presenting with reading difficulties.5 RPE mottling in younger subjects progresses to a bull’s-eye maculopathy (BEM) and, later, macular atrophy. Some patients have typical features of RP (see Chapter 44). Patients have a mild to moderate reduction in visual acuity, except when associated with RP where markedly reduced central vision is common.
Best’s disease (vitelliform macular dystrophy)
Clinical and histological findings
Best’s disease (BD) is an autosomal dominantly inherited macular dystrophy with a round or oval yellow subretinal macular deposit that is highly autofluorescent on AF imaging (Fig. 46.5). Although most gene carriers show a reduced light rise on EOG, the retinal phenotype is variable. Some gene carriers have a completely normal fundus. The phenotype is classified into five stages (Table 46.2):
Stage 0: pre-vitelliform − a normal fundus in an asymptomatic gene carrier with an abnormal EOG.
Stage II: classical “egg yolk” macular lesion (Fig. 46.6A). FFA shows a corresponding area of blocked choroidal fluorescence (Fig. 46.6B). This appearance is usually seen during the first or second decades, often with near normal visual acuity.
Stage IIa: the “egg yolk” begins to break up secondary to resorption of the yellow material lying between the RPE and sensory retina, usually associated with noticeable visual impairment (Fig. 46.7).
Stage III: pseudohypoyon − part of the lesion is resorbed leaving the appearance of a “fluid level” at the macula.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


