Inflammatory Choroiditis
Jose S. Pulido
C. Richard Blake
There are a group of ocular diseases that appear to be inflammatory reactions in the choroid. The cause of the inflammatory reaction is unknown in all of these entities. Some of these entities are considered infectious, but there has been no histologic evaluation to show the presence of infectious agents that would be required to confirm that is the case. Another possibility is that they are immunologic in origin but there also is no definite confirmatory determination of this.
The reason the diseases are considered infectious is because they appear to cause acute visual loss and some are associated with white blood cells in the vitreous or a retinal vasculitis. In addition, the lesions may crop up and have an acute, chronic, or semiacute course similar to systemic infectious diseases. Many of these entities affect persons in the third to fifth decade of life, which is when many of the immunologic reactions occur elsewhere. It may be that there is an initial infectious agent that in a host with certain predisposing characteristics triggers an immunologic reaction, or it could be that the agent stays indefinitely in the host causing a host-induced reaction.
On physical examination, the patients may have iritis, vitreitis, or retinitis, although in many of the choroiditic entities they may not manifest these findings. Because the choroid or the retinal pigment epithelium is affected, they may show a serous retinal detachment that presents as a blister of fluid that is loculated or as an exudative retinal detachment with shifting subretinal fluid. Acutely there may be a gray-yellow plaque of varying size. It may then resolve with only mild almost imperceptible pigmentary changes or there can be dense white chorioretinal scarring. Because the photoreceptor/retinal pigment epithelial complex is affected, many note photopsias or colored swirling lights and also they may have visual field defects.
It is important to determine carefully that the disease is truly an inflammatory disease and not a known infectious or neoplastic disease. Infectious choroiditides have to be considered in all circumstances of inflammatory choroiditis. A specific test for Treponema pallidum, for instance, the fluorescent treponema antibody-absorbed (FTA-ABS) test or the microhemaglutination-treponema pallidum test (MHA-TP) should be performed to determine if the choroiditis is caused by syphilis. Other diseases, including tuberculosis, cat-scratch disease, coccidioidomycosis, West Nile encephalitis, and nocardiosis among others need to be considered. In addition, certain noninflammatory diseases, for instance, central serous chorioretinopathy may have an acute time course similar to that of an inflammatory condition and eclampsia, thrombotic thrombocytopenia purpura, and disseminated intravascular coagulation may also mimic an acute inflammatory choroiditis.
WHITE DOT SYNDROMES
One group of these inflammatory choroiditides are subclassified under the auspices of white dot syndromes. These tend to have multiple lesions. The patients tend to be young women and in addition to visual loss they may have photopsias. The photopsias are caused by photoreceptor damage during acute episodes of choroiditis. This group of diseases consists of multiple evanescent white dot syndrome (MEWDS), acute zonal occult outer retinopathy (AZOOR), acute annular outer retinopathy (AAOR), punctate inner choroiditis (PIC), multifocal choroiditis, acute posterior multifocal placoid pigment epitheliopathy (APMPPE), birdshot chorioretinopathy, and diffuse subretinal fibrosis. Presumed ocular histoplasmosis syndrome is sometimes included in this group although in direct contradistinction to the other diseases, the patients are usually asymptomatic until they develop a choroidal neovascular membrane.
MULTIPLE EVANESCENT WHITE DOT SYNDROME
HISTORY AND CLASSIC FINDINGS
MEWDS was first described by Jampol and associates1 and in the same year by Takeda and coinvestigators.2 The first cases involved acute unilateral visual loss and scotomas in 12 young women and 3 men. All had multiple white lesions at the level of the retinal pigment epithelium and approximately half had a previous flu-like syndrome. Macular granularity was seen commonly in these cases.
EPIDEMIOLOGY
PATHOPHYSIOLOGY
The etiology of this form of white dot syndrome is unknown. There is a history of a precedent upper respiratory tract infection in 25% to 50% of cases.5 It could be that in certain predisposed individuals, exposure to a certain infectious organism can trigger an immune reaction to the choriocapillaris or the overlying retinal pigment epithelium. There is one case of murine typhus associated with MEWDS-like fundus findings.6 There is also a case with MEWDS-like findings that occurred after hepatitis B vaccination.7
INITIAL CLINICAL FINDINGS
Presenting Symptoms
The most common presenting symptoms are unilateral loss of vision and photopsias. These photopsias are described as flashing colored lights. Occasionally they may complain of a unilateral blind spot. Rarely do they have bilateral symptoms.
There may be a history of a preceding upper respiratory tract infection. Usually this is not volunteered by the patient because if it had been present, it was mild and so the patients do not consider it related to the present problem.
Signs
VISION
The vision is usually only mildly diminished although the vision may range from 20/20 to 20/400. Color vision may be slightly or moderately diminished as well.
PUPILS AND FIELDS
The visual fields are variable but most often they show an enlarged blind spot. There may be an afferent papillary defect as well.
BIOMICROSCOPY
Iritis is rare but has occasionally been described. Vitreitis is more common although if it is present, it tends to be very mild.
FUNDUS FINDINGS
The classic fundus findings are small (500 μm), ill-defined, and sometimes difficult to see grayish-white dots that are dispersed throughout the posterior pole. These spots rarely may become confluent and if this occurs to spots that are around the disc, it may present as a case of giant blind spot syndrome or of an entity that probably is a variant of MEWDS called AZOOR.8 In addition to the dispersed small spots, there is a fine granularity of the fovea. This finding is pathognomonic for this disease, and it can exist in the absence of the white dots (Figs. 1 and 2).
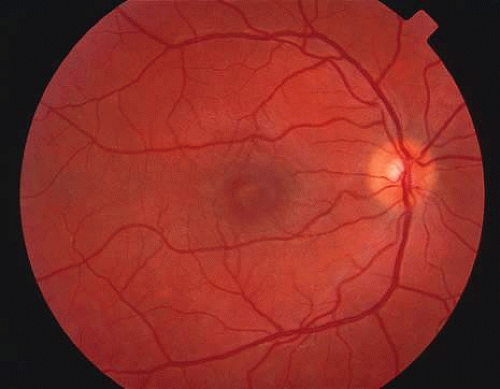 Fig. 1. Color fundus photograph of a patient with multiple evanescent white dot syndrome. Notice that the foveal spot and irregularity is much easier to detect than the subtle white dots. |
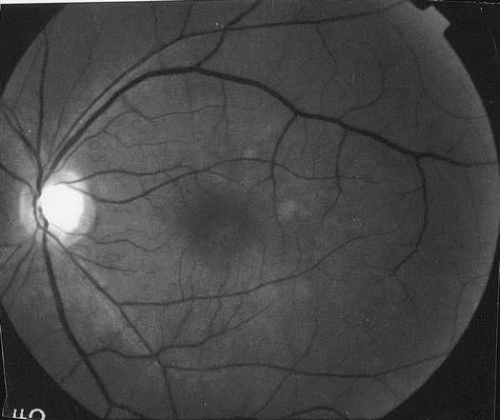 Fig. 2. Red free black and white photograph of a different patient with MEWDS. This technique accentuates the white dots but does not show the foveal changes as well. |
Usually there is resolution of the fundus findings, although careful fundus evaluation sometimes shows some residual fine retinal pigment epithelial mottling. In rare cases, there may be long-standing pigment clumping with surrounding pigment atrophy similar to the lesions seen in multifocal choroiditis or punctate inner choroidopathy although that has been contended. The foveal granularity may take longer to resolve than the white spots.
There may be mild optic disc swelling. This may lead someone unfamiliar with this disease to consider that the cause of visual loss is optic neuritis. There also may be mild retinal phlebitis that may be more easily seen by fluorescein angiography than by ophthalmoscopy. Choroidal neovascularization rarely develops.9
ANCILLARY TESTING
Fluorescein
The fluorescein angiograms of the white dots show a fine halo appearance in a wreath-like swirl configuration to some of the white dots, and this halo diffuses in the later stages of the angiogram, first developing a stippled pattern of fluorescence and then becoming a diffuse white spot that leaks only mildly. There may be leakage of fluorescein from the optic nerve and occasionally, there may be late staining of retinal vessels because of a mild vasculitis (Figs. 3 and 4).
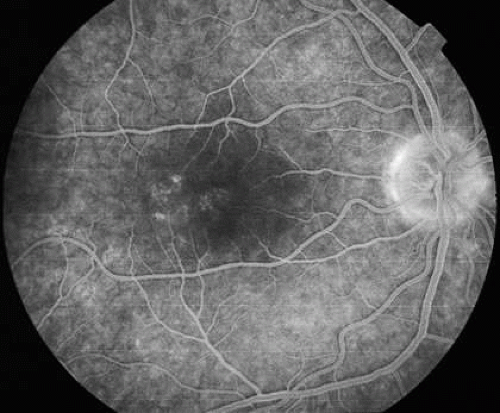 Fig. 3. Venous phase fluorescein angiogram of fundus shown in Figure 1. It demonstrates the hyperfluorescent dots coalesced near the fovea and in a subtle wreath-like fashion elsewhere. |
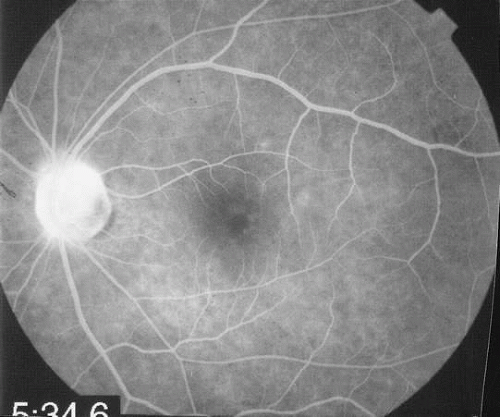 Fig. 4. Recirculatory state fluorescein angiogram of fundus shown in Figure 2. It demonstrates the leakage from the disc and the fluorescence of the white dots is more evident. |
Indocyanine Green
In the early stages of the angiogram, the indocyanine green (ICG) angiogram may not show any distinct abnormalities. In the late phases, ICG angiography can show more lesions than can be seen by ophthalmoscopy or by fluorescein angiography. These lesions tend to block or show absence of fluorescence.10,11 In addition, there can be many hypofluorescent lesions around the disc. Sometimes the spots are so extensive that they become confluent. It is thought that it is a confluence of these small lesions that cause the blind spot enlargement seen on visual field testing. At the peripheral edge of the confluent spots however, it is still possible to see single spots that are characteristic with the stippled halo-like fluorescence.12 The hypofluorescent spots noted by ICG can last longer than the symptoms, or ophthalmoscopic or fluorescein angiographic findings.13
Electroretinography
Electroretinography (ERG) shows a diffuse loss of the early receptor potential and a decrease of the a wave, indicating that the disease appears to affect the retinal pigment epithelium-photoreceptor complex.14 Multifocal ERG shows reduced a and b waves in the areas that correspond to the visual field scotomas as well as showing areas of dysfunction where the visual field does not seem affected.15 In addition, the reduced amplitudes may be noted months after the visual fields and visual acuity have returned to normal. In patients with acute idiopathic blind spot enlargement (AIBSE) syndrome, a disease that may be related to MEWDS, the multifocal ERG is similarly affected in the peripapillary area.16 These patients may actually have had MEWDS in later stages of resolution because the white lesions may resolve much earlier than the scotomatous changes.17 Electrooculography may also show a diminution of the dark peak to light trough ratio.
Laboratory Tests/Immune Testing
There is a small study that showed a higher incidence of the HLA B51 haplotype in persons with MEWDS compared to the normal population. There is a relative risk of 5.86. This study needs to be confirmed in a larger cohort.18 It is interesting to note that this haplotype is also seen more frequently in patients with Behçet’s disease, but in Behçet’s disease the relative risk is almost twice as high as in MEWDS.
Testing for immunoglobulin staining in patients with different causes of outer retinopathy have not shown reactivity with the retina. This does not necessarily mean that the disease does not involve an immune response to the retina. It means that the immune reaction does not involve autoantibodies. Cell-mediated immunity may still be present.19
Differential Diagnosis/Mimics
The differential diagnosis tends to involve the other causes of the white dot syndromes. Most commonly the diagnoses that are most often confused with MEWDS are APMPPE, birdshot retinochoroidopathy, multifocal choroiditis, and retinal pigment epitheliopathy. In addition, acute macular neuroretinopathy and diffuse unilateral subacute neuroretinitis are sometimes considered in the differential diagnosis. None of the other white dot syndrome diseases have the peculiar foveal granularity that is seen with MEWDS.
APMPPE is usually a bilateral disease that affects both young men and women. The creamy lesions tend to be larger than those seen in MEWDS, and fluorescein angiography reveals classic early blocked fluorescence and late staining of the lesions.
Birdshot chorioretinopathy usually affects older women between 40 and 60 years of age. It causes deep creamy lesions the borders of which are sometimes difficult to discern. In the late phases of fluorescein angiography some of the lesions show hyperfluorescence while others do not show up on the angiograms. The optic nerve tends to be swollen and shows leakage by fluorescein angiography. Cystoid macular edema may be present.
Many times the lesions in multifocal choroiditis show lesions in different stages of scarring. Choroidal neovascularization, bilaterality, and recurrences are common.
Acute retinal pigment epitheliopathy shows a few small spots that are noted to have pigment clumping in their center with surrounding hypopigmentation. Fluorescein angiography shows window defect with a central spot of blocked fluorescence. The electroretinogram is unaffected.
Acute macular neuroretinopathy tends to be unilateral. There is a mild diminution of vision associated with a reddish discoloration centered under the fovea. This pattern is most easily seen with red free photography.
Diffuse unilateral subacute neuroretintis is caused by a nematode. It can be associated with creamy-white small lesions in the macula. This is a progressive disorder unless the nematode is identified in the subretinal space and treatment is given.
There are three cases of two men and one woman all in their 50s who had whitish spots that were consistent with those seen in MEWDS. The age was atypical and there was progressive vision loss in these cases. These patients had primary intraocular large-cell (non-Hodgkin’s) lymphoma.20 In no case were there the pathognomonic foveal changes seen in MEWDS.
Natural History
The symptoms usually resolve within 6 to 15 weeks although some may have the blind spot enlargement and photopsias for up to 1 year after the initial presentation.5,21 Recurrence is rarely noted and choroidal neovascularization is also a rare occurrence. Punched-out lesions, similar to those seen in multifocal choroiditis, can rarely be seen in a few cases.
Treatment and History with Treatment
Treatment is not indicated because the disease spontaneously improves. Laser photocoagulation or possibly photodynamic therapy may be of help in cases that develop choroidal neovascularization.
ACUTE IDIOPATHIC BLIND SPOT ENLARGEMENT SYNDROME
AIBSE is probably a variant of MEWDS. The patients tend to be women between the ages of 20 and 50 years. There may be no to moderate vision loss. An enlarged blind spot is invariably present. The patients have photopsias as well. Although some cases have the white dots similar to MEWDS in the retina, others do not and that is why it is argued that AIBSE is a different disease. In addition, some cases do not have the macular pigment granularity classically seen in MEWDS.22 Some may develop recurrences. It may be that AIBSE are actually cases of MEWDS that have had resolution of the fundus lesions.
ACUTE ZONAL OCCULT OUTER RETINOPATHY
AZOOR was the term used by Gass and colleages23 to describe the ocular findings in a disease noted more commonly in women than men (73% vs. 27%). The patients tend to be young and myopic (66%), and the disease starts unilaterally in 60% of the cases with the fellow eye ultimately becoming involved in 60% of the unilateral cases. There can be a delay of up to 15 years between the first and the second eye becoming involved, although the mean time for the involvement of the fellow eye was 50 months. Bilateral disease is more common in males than in women (100% vs. 68%). Recurrences occur in approximately one third of cases and the mean time to each recurrence was 52 months. An antecedent flu-like illness was noted in 20% of cases and 13% had headaches prior to development of the ocular findings. Approximately 25% of patients had a history of a systemic autoimmune disease.
Most patients develop acute visual loss followed by stabilization after 6 months. Only a rare patient has stepwise progression of visual loss. Patients note the visual loss as scotomas that involve one or more parts of the visual field with the most common finding being an enlarged blind spot. The vast majority of patients (88%) notice photopsias that may be colored or noncolored. Most patients have visual acuity of 20/40 or better on initial evaluation, and the majority retain 20/40 or better at the time of the final evaluation. Vitreous cells are present in approximately one half of the cases and approximately 10% of eyes were noted to have retinal vascular sheathing. The eyes with vitreous cells may develop focal areas of mottling of the retinal pigment epithelium that is similar to the mottling seen in mild cases of retinitis pigmentosa although many eyes appear to have a normal fundus. Rarely, patients may develop chorioretinal scars that appear similar to those seen in multifocal choroiditis. Fluorescein angiography in the acute phase is usually unremarkable. In the chronic phase there may be mottling fluorescence at the level of the retinal pigment epithelium. An abnormal full-field electroretinogram was noted in more than 60% of the patients. The multifocal electroretinogram may also be abnormal.24 The differential diagnosis in this group of patients includes the white dot syndrome diseases, diffuse unilateral subacute neuroretinitis, retinitis pigmentosa, carcinoma associated retinopathy, and syphilis.
ACUTE ANNULAR OUTER RETINOPATHY
AAOR may be a variant of AZOOR.25,26 The first case was reported by Luckie et al.27 The second case was reported by Gass and Stern.28 These cases are characterized by visual loss and visual field loss in young adults associated with a demarcation ring of gray-white outer retinal coloration that separates the area of the retina with visual field loss and normal retina. There are no discernible retinal findings within the annulus of whitening. Usually the diametric center of the annulus was the optic disc. With time the grayish discoloration abates and there is some thinning of the retinal vessels within the area of affected retina. In some cases there was mottling of the retinal pigment epithelium. There were no vitreous cells noted and the fellow eye has not been affected in the few cases that have been reported. Fluorescein angiography may be normal initially and later there may be mottled fluorescence at the level of the retinal pigment epithelium.
There appears to be a group of diseases that involve the retinochoroidal junction and these include multifocal choroiditis, acute macular neuroretinopathy, AIBSE, PIC, AZOOR, AAOR, and MEWDS. Because there have been some patients that have two of these diseases together or in progression, Gass and Stern28 have suggested they are all related. Others have seen cases of overlap between these syndromes as well.29 AZOOR disorders are characterized by rapid visual field loss and electroretinographic findings that show fundus involvement far in excess of that seen by fundoscopy.30 All of these patients tend to have photopsias as well. There is one case of AZOOR associated with a relapsing-remitting acute case of cervical myelitis.9,19,31,32 Many cases have a history of an antecedent viral illness, and it may be that these diseases are caused by a transient viral infection of the photoreceptors and/or the retinal pigment epithelium or there may be an immunologic reaction that affects these layers following the viral infection. Treatment with antiviral agents does not appear to be efficacious and many of these diseases are self-limited.
ACUTE POSTERIOR MULTIFOCAL PLACOID PIGMENT EPITHELIOPATHY
HISTORY AND CLASSIC FINDINGS
APMPPE was first described by Gass.33 He described three patients between the ages of 19 and 22 years in whom both eyes were affected. There were creamy white spots that were associated with mild to marked visual loss. The lesions typically blocked early and stained late by fluorescein angiography, and usually there was resolution with only mild pigment clumping. Recurrences are rare.
EPIDEMIOLOGY
Most cases affect young persons both men and women between the ages of 20 and 40 years. There has been an association with other diseases as noted below.
PATHOPHYSIOLOGY
The fact that there have been cases after known exposure to other diseases or antigens means that this may be a focal response to the systemic inflammatory process. There is an association with cerebral vasculitis, which also supports this possibility. There may be an HLA predisposition that would lend credence to the idea that if a person with a certain immunogenetic haplotype is exposed to certain antigens, he or she may develop APMPPE as a response. Approximately 25% to 40% of patients report a previous prodromal viral illness. It may be that there is a choroidal vasculitis with a secondary reaction in the overlying retinal pigment epithelium.
INITIAL CLINICAL FINDINGS
Presenting Symptoms
SIGNS
The patient may present with complaints of photopsias, scotomas, and possible vision loss. Both eyes tend to be affected although they can be asymmetrically affected.
VISION
The vision is variable and depends on whether the lesions are under the fovea. The vision may be normal or reduced to 20/200 or worse.
PUPILS AND FIELDS
There are visual field defects that correspond to some of the fundus lesions. There can be an afferent papillary defect if both eyes are very asymmetrically affected.
BIOMICROSCOPY
There may be a mild iritis although most cases do not show it. More commonly there may be a mild vitreitis.
FUNDUS FINDINGS
Acutely, there may be creamy white lesions in the posterior pole. These lesions tend to be deep and poorly demarcated. Sometimes these lesions may become confluent. The lesions clear by losing their yellow-white opacification and clearance occurs from centrally to the periphery. With time the lesions resolve leaving mottling of the retinal pigment epithelium (Figs. 5, 6, and 7).
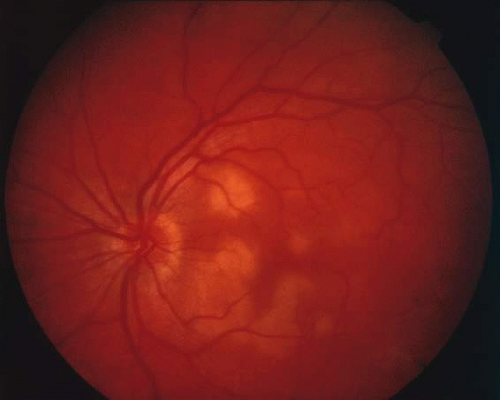 Fig. 5. Fundus photograph showing the acute creamy lesions of a recent case of acute multifocal posterior placoid pigment epitheliopathy. |
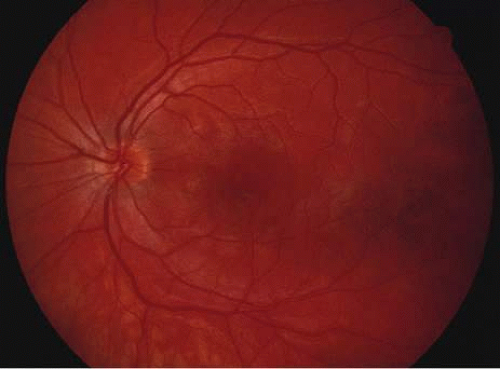 Fig. 6. Fundus photograph of the same case as in Figure 5. The findings at 1 month after the initial presentation now shows mild pigment clumping and mild retinal pigmentary atrophy. |
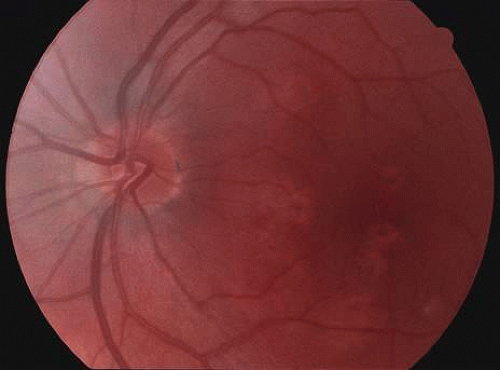 Fig. 7. Fundus photograph of the same case as in Figure 5. The findings at 7 months following initial presentation show more pigment clumping and retinal pigment atrophy in small circular lesions. |
SYSTEMIC ASSOCIATIONS OF ACUTE POSTERIOR MULTIFOCAL PLACOID PIGMENT EPITHELIOPATHY
There have been multiple reports of a cerebral vasculitis in association with APMPPE. These patients may have a cerebral spinal fluid pleocytosis. In patients with APMPPE who complain of headaches or have other central nervous system findings, magnetic resonance imaging should be strongly considered and the patient should be placed on high doses of corticosteroids. The interval between the APMPPE and the onset of symptomatic cerebral vasculitis may be up to 3 months.
Mumps have been associated with APMPPE as well as immunization with hepatitis B virus vaccine. In the case of the immunizations, the APMPPE occurred after the booster injection. There has been a case of APMPPE after streptococcal infection.34
ANCILLARY TESTING
Fluorescein
The classic findings in the acute cases are lesions of variable size that are hypofluorescent in the early phase of the fluorescein angiogram. In the late phases of the angiogram there is hyperfluorescence of the lesions. In the late phases, there also may be leakage from the disc as well, and there also may be staining of the retinal vessels because of the vasculitis. With resolution of the disease, there still may be a mottled pattern of fluorescence correlating with the areas of pigment clumping and pigment atrophy (Fig. 8).
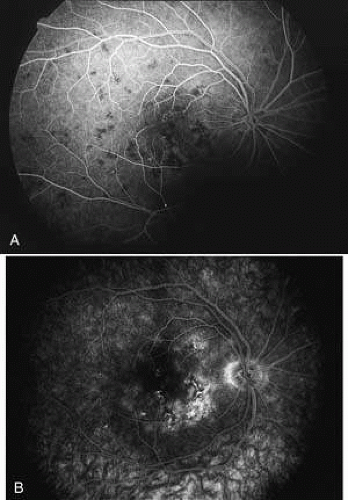 Fig. 8. A. Early fundus angiogram showing hypofluorescence of the acute posterior multifocal placoid pigment epitheliopathy (APMPPE) lesions. B. Later stage of the angiogram showing staining of the APMPPE lesions. |
Indocyanine Green
ICG angiography shows an absence of fluorescence in the early as well as in the late phases of the angiogram. This is consistent with the possibility that the disease is caused by a choroidal occlusive vasculitis. There may be more lesions than seen ophthalmoscopically or by fluorescein angiography. In addition, there may be the development of new lesions in the first few weeks that can be seen by indocyanine green that cannot be seen by fundoscopy.35 This appears to imply that the disease is first a choroidal vasculitis, and in the more severe cases it can then affect the overlying retinal pigment epithelium and outer retina.
Other Diagnostic Findings
The electroretinogram and electrooculography may both be reduced in APMPPE. There may also be a prolonged dark-adaptation, color match abnormalities, and an abnormal Stiles-Crawford effect. These abnormalities resolve after 1 year. By fundus reflection densitometry, there is decreased cone pigment in the acute phase and pigment regeneration is prolonged; these findings also resolve with time.36,37
Laboratory Tests/Immune Testing
The HLA B7 and the HLA DR2 haplotype were seen more often in patients with APMPPE in one report, but that has not been confirmed.
Differential Diagnosis/Mimics
Although the differential diagnosis includes all other white dot syndromes, the most common diseases that closely resemble APMPPE are serpiginous choroiditis, MEWDS, birdshot choroidopathy, diffuse metastatic cancerous lesions, non-Hodgkin’s lymphoma, and Pneumocystis choroiditis. Subacute sclerosing panencephalitis caused by chronic measles infection can have similar fundus findings. If the patient is older than the normal group of patients, if the lesions are not exactly typical, or if there are systemic findings, then a complete systemic work-up should be considered.
Serpiginous choroiditis is associated with recurrences that are rare or probably do not occur with APMPPE. In addition, multiple separate lesions are rare in serpiginous choroiditis but common in APMPPE. By contrast, choroidal neovascularization is rare in APMPPE but common in serpiginous choroiditis. Both tend to be bilateral but there is asymmetry in the time course of the lesions in serpiginous choroiditis whereas in APMPPE the lesions of both eyes tend to follow a similar time course. Finally, patients with serpiginous choroiditis tend to be middle-aged compared to young adults in APMPPE.
Natural History
Usually, the lesions resolve leaving only mild pigment mottling and the vision also improves. Resolution begins after a few weeks and may continue for up to 1 year. Choroidal neovascularization may occur with APMPPE but is a rare finding. Sometimes there may be significant pigment changes and even mild chorioretinal scarring. In cases where the pigmentary changes are under the fovea, visual improvement may be limited. In one series, 11% had visual acuity of 20/200 or worse on follow-up, although another series had 94% of affected eyes having a final visual acuity of 20/30 or better.38 Usually if there is marked chorioretinal scarring, serpiginous choroidopathy should be considered a possible diagnosis.
Treatment and History with Treatment
Treatment is only indicated in cases with concomitant cerebral vasculitis or in cases where the vision is markedly diminished. Corticosteroids are used in those cases. It is not certain that corticosteroids affect the visual outcome but it appears to affect the course of the cerebral vasculitis. In cases where the cerebral vasculitis does not appear to improve or in cases where there is marked bilateral visual loss that is unresponsive to corticosteroids, consideration for using cyclosporine should be given.
PRESUMED OCULAR HISTOPLASMOSIS
HISTORY AND CLASSIC FINDINGS
Presumed ocular histoplasmosis (POH) is probably caused by dissemination of the histoplasma capsulotum after acute exposure to the organism. It is characterized by punched-out chorioretinal lesions, peripapillary scarring, and choroidal neovascular membranes, with no vitreous or anterior chamber inflammation.
This disease is seen most often in people who come from the Ohio-Mississippi River Valley area where the fungus is ubiquitously found. Most often patients present in their fourth and fifth decade of life because of the development of a choroidal neovascular membrane. There may be a genetic predisposition and patients that have the HLA-B7 or the HLA-DR2 haplotype may be more susceptible to developing the ocular form of the disease.
It is hypothesized that after acute exposure to the organisms by inhalation in susceptible persons, there may be a pulmonary infection associated with symptoms of a lower respiratory tract infection including mild fever and a cough. This elicits a granulomatous response and the organisms are probably destroyed by this cell-mediated response.
As opposed to the inflammatory choroiditides, POH tends to be asymptomatic unless there is a choroidal neovascular membrane that is causing visual symptoms. There have been a few cases reported that appear to show that there can be symptomatic exacerbations of the inflammatory component of the choroidal scars. These cases are rare and they appear to respond to corticosteroids. There are also rare cases of intrascar neovascularization that may imitate the exacerbation of inflammation. It is hard to distinguish intrascar neovascularization from a true exacerbation of inflammation because they both show subretinal fluid, the patient is symptomatic in both, and the lesions respond to corticosteroids many times in both cases or at least temporarily in cases of intrascar neovascularization. If there is subretinal hemorrhage, the cause of the symptoms is neovascularization. Once the neovascularization extends to the edge of the scar, then laser photocoagulation or photodynamic therapy should be considered. These intrascar neovascularizations are different from classic cases where the neovascularization begins at the edge of the scar.39
BIOMICROSCOPY
Rarely, there are some cases of documented choroiditis associated with histoplasmosis. There are however rare cases and if there is choroiditis, other diagnoses, especially multifocal choroiditis, should be considered.
MULTIFOCAL CHOROIDITIS
HISTORY AND CLASSIC FINDINGS
Multifocal choroiditis and panuveitis are also considered in the differential diagnosis of inflammatory white dot syndromes. This disease is usually seen in middle-aged women who present with visual loss with the presence or absence of photopsias, vitreitis, and sometimes iritis, multiple chorioretinal scars that are of variable size but usually no larger than 1 mm unless they coalesce. The lesions can develop choroidal neovascularization.
EPIDEMIOLOGY
Women are affected approximately fourfold more often than men.40 The mean age of the patients at the time of initial presentation is approximately 35 years of age.
Signs
Disease tends to be bilateral on presentation in approximately 80% of the cases. There can be significant asymmetry between the two eyes at the initial presentation.
Vision
On presentation, the initial visual acuity of affected eyes tends to be approximately 20/50.
Pupils and Fields
There may be enlarged blind spots secondary to confluence of lesions around the disc. In addition there can be scotomas associated with the choroidal lesions and these can enlarge if there is a choroidal neovascular membrane around the scar. The size of the visual field defects tend to stay constant as opposed to visual field defects associated with PIC or MEWDS that can become smaller.
Biomicroscopy
MULTIFOCAL CHOROIDITIS
Anterior Segment Findings
If there are corneal infiltrates then there should be consideration that the multifocal choroiditis is caused by Epstein-Barr virus. Syphilis and sarcoidosis should be considered if there is a deep interstitial keratitis in the presence of a choroiditis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


