Chapter 27 Histiocytic, hematopoietic and lymphoproliferative disorders
1. Langerhans’ cell histiocytosis (LCH, histiocytosis X): abnormal histiocytes are derived from dendritic (granular) Langerhans’ cells, involved in antigen presentation.1,2 These cells have characteristic inclusions visible on electron microscopy.
2. Non-Langerhans’ cell histiocytosis results from proliferation of another class of dendritic histiocytes, the dermal dendrocytes, lacking inclusion granules. These are responsible for juvenile xanthogranuloma.
The macrophage system, a third type of histiocytic cell, gives rise, through a polyclonal proliferation, to sinus histiocytosis, also known as Rosai-Dorfman disease.3 These three histiocytic disorders will be discussed in this chapter.
Langerhans’ cell histiocytosis (histiocytosis X)
Langerhans’ cell histiocytosis (LCH) is predominantly a childhood disorder with a peak incidence from 1 to 3 years of age and an annual incidence of 4−5 per million.4 Boys are more frequently affected, though female patients may be younger, with more widespread disease.5 Langerhans’ cells are dendritic histiocytes with minimal phagocytic capacity, involved in immune surveillance. Normally situated at the junction of dermis and epidermis, they migrate to regional lymph nodes after antigen encounter, where they participate in antigen presentation.6
LCH lesions are destructive and space-occupying producing a clinical picture which varies with the site and the tissue involved. In children, the disease most commonly affects bones, especially those involved in hematopoiesis, and skin. Lesions may occur in the other organs containing histiocytes and macrophages such as the spleen, liver, lymph nodes, and lung. Data from the early 1990s suggested a monoclonal neoplastic origin to this disorder6 but further investigation has not shown consistent genomic aberrations.7 The exact etiology of this disorder remains obscure. Langerhans’ cells in the skin multiply from single progenitor cells in embryonic development. LCH cells may react to the local cellular environment including T cells and cytokines in an abnormal manner.8 The result is highly variable, possibly depending on cell mutations, interaction with immune surveillance and the site of origin of the affected cell.
Alfred Hand9 first reported polyuria, exophthalmia and skull destruction in a 3-year-old. Subsequently, conditions describing the spectrum of histiocytic disease were given eponymous names; eventually, three different clinical disorders were described:
1. Eosinophilic granuloma, typically affects children aged 4–7 years. The lesions are confined to bone.
2. Hand-Schuller-Christian disease affects younger patients and is more widespread and aggressive with multifocal lesions at the skull base causing the triad of diabetes insipidus (from infiltration of the hypothalamus and/or posterior pituitary), exophthalmos, and bony defects of the skull.
3. Letterer-Siwe disease − often affects children under 2 years of age causing multisystem involvement including cutaneous, lymph node, visceral, ocular, and orbital disease. This is the most aggressive end of the LCH spectrum, and is frequently fatal.
Since there is significant clinical overlap and identical histopathology in all three groups, Lichtenstein10 called the whole group “histiocytosis X” to emphasize the common cell of origin and the unknown etiology. The term “Langerhans’ cell histiocytosis” replaced histiocytosis X in 1987, differentiating conditions in which the abnormal histiocytes are derived from the Langerhans’ cell from the other histiocytic disorders. LCH has been subdivided clinically into:
1. Single system disease, which may be limited to a single site (unifocal, usually corresponding to eosinophilic granuloma), or involve multiple sites (multifocal, corresponding to Hand-Schuller-Christian disease)
2. Multisystem disease (systemic, previously known as Letterer-Siwe disease).
Ophthalmic involvement
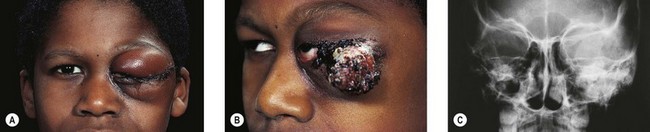
The most common ophthalmic presentation of LCH is orbital involvement (Fig. 27.1),5,11 but disease of the globe and brain may also lead to ophthalmic consultation. Intraocular lesions, with infiltration of the uveal tract, are seen most frequently in infants with disseminated LCH.12 Intracranial involvement may cause visual field defects due to infiltration of the optic nerves, chiasm, or tracts. Cranial neuropathy13 and raised intracranial pressure are occasional presentations.
Orbital involvement
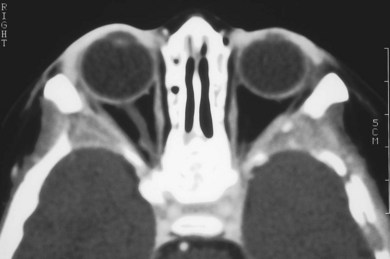
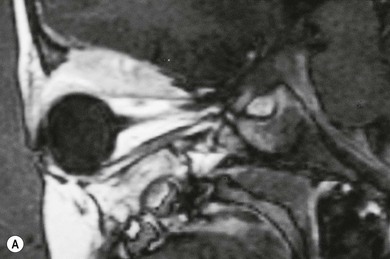
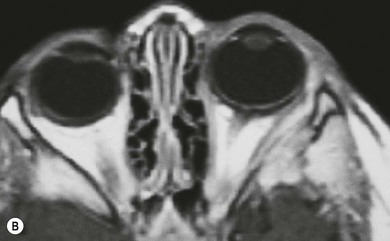
The orbit is involved in 20% of cases of LCH,11 usually with the localized form of the disease (eosinophilic granuloma); it is rare in patients whose disease is limited to soft tissue, suggesting that the lesion usually arises in the bone.11 They are usually situated superotemporally, with a predilection for the frontal and parietal bones and the greater wing of the sphenoid.14,15 Radiologically, the lesions are lytic, with a soft tissue component causing expansion of the surrounding tissues (Figs 27.1 to 27.4). Occasionally, lytic bony lesions may be seen radiologically in the absence of any clinical signs.
Clinical features
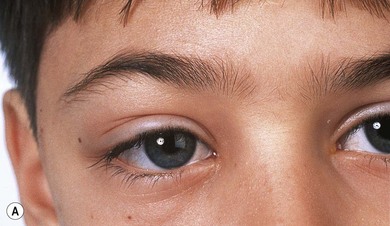
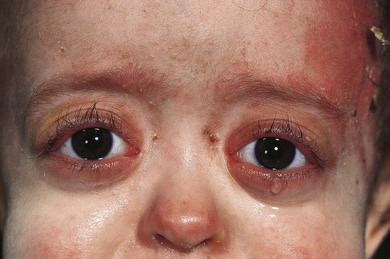
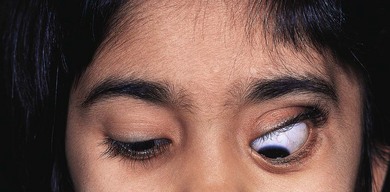
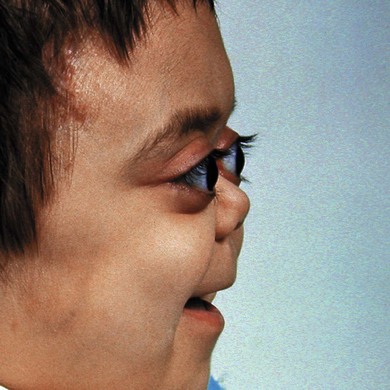
The usual presentation is with unilateral or bilateral proptosis.5 Rarely, the proptosis may be extreme causing luxation of the globe.16 Less commonly, the presentation is with isolated orbital involvement in a previously healthy child, in which case the disease is usually unilateral. Initially, the course may be evanescent and relapsing. An isolated lesion of the superior orbital wall may present with unilateral ptosis or inferonasal globe displacement. The lesions, if superficial, are generally soft to palpation. Optic nerve compression and cranial nerve palsies are rare, but may be seen with extensive orbital involvement.11 Skin tethering (Fig. 27.5), erythema, and erosion may occur (see Fig. 27.5). Visual loss may be caused by optic nerve compression,11 optic atrophy due to chronically raised intracranial pressure,11 chiasmal disease, or intraocular infiltration.12,17,18 Chronic disseminated LCH (Hand-Schuller-Christian disease) may present with polyuria and polydipsia, and can be associated with growth, thyroid, and gonadotrophic hormone deficiencies.
Investigation
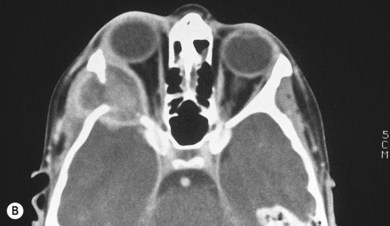
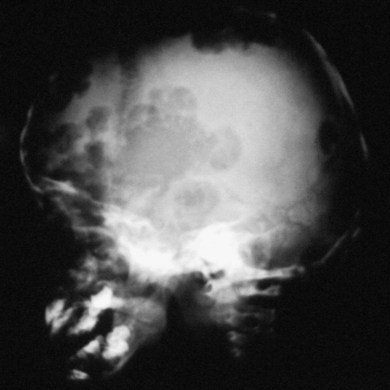
In most children with orbital disease, plain X-rays (Fig. 27.6) will demonstrate a lytic bone lesion. Computed tomography (CT) scan demonstrates an enhancing mass often with a low density center; together with magnetic resonance imaging (MRI) this will delineate the extent of intraorbital bone or intracranial involvement (Fig. 27.7). On MRI, bony lesions are typically hypointense on T1-weighted images with intermediate to high signal intensity on T2-weighted images. Most lesions show moderate to intense contrast enhancement. MRI is preferable for evaluation of intracranial extension.18 Orbital lesions usually remain extraconal but may spread into the muscle cone.
The diagnosis is confirmed by histologic examination of involved tissue. In children with multisystem disease, an accessible site such as skin or a peripheral bone should be biopsied; in cases with solitary orbital involvement, orbital biopsy is necessary. Fine needle aspiration may be useful in these circumstances.19 Children who present to the ophthalmologist should be referred to a pediatric oncologist to determine if there is any systemic involvement. Further investigations may include chest X-ray, skeletal survey, lung and liver function tests, and specific gravity of early morning urine.
Histopathology reveals granulomatous infiltration consisting of histiocytes and multinucleated lipid-laden giant cells, eosinophils (particularly numerous in single site bone-based lesions or “eosinophilic granulomas”), lymphocytes, plasma cells. and neutrophils. Electron microscopy demonstrates typical Langerhans’ granules, also known as Birbeck or racket bodies, in about 50% of cases,20 indicating that the proliferating histiocytes are derivatives of the Langerhans’ cells, part of the mononuclear–phagocyte system.21 Immunohistochemical techniques may be helpful to secure the diagnosis.2
Management and prognosis
Close dialogue between the treating ophthalmologist and oncologist in the management of LCH with orbital involvement is essential.22 Advice from other specialists such as ENT and orthopedic surgery may be needed.
The management of orbital lesions depends on whether there is single system or multisystem disease.23 Conservative management with careful observation may be justified in patients with single site orbital involvement, where complete spontaneous resolution after incisional biopsy24 or even fine needle aspiration has been reported. Usually, patients with a single orbital lesion are treated by biopsy and curettage resulting in resolution.11 Intralesional steroids may be used to hasten remission11 or reduce pain. If there is marked proptosis or optic nerve compression, a short course of systemic steroids or radiotherapy may be used to induce remission. A radiation dose of 500–600 cGy is usually sufficient. The total dose should not exceed 1000 cGy because of the risk of radiation-induced malignancies in later life. Cosmetically disfiguring lesions of the orbital wall may be removed surgically with curettage. Orbital bone involvement frequently results in arrested growth of the walls with shallowing. This is not related to the use of radiotherapy (Fig. 27.8).
Children with single system disease, for example of the bone, have a good prognosis.25,26 The prognosis is poor in multisystem or visceral disease, especially if there is infiltration and failure of key organs such as the bone marrow, liver, and lungs, which may be fatal.7 Children under the age of 2 years have a mortality rate of 55–60%; death is rare after the age of 3 years. Although some authors have stressed age as the most important prognostic factor, it is really the tendency of infants to develop multisystem disease rather than their age which dictates the poorer prognosis of children aged 2 years and under.23,27 Response to the initial treatment of multisystem LCH is a good predictor of eventual outcome.23,28
Non-Langerhans’ cell histiocytosis
Juvenile xanthogranuloma
Juvenile xanthogranuloma (JXG) is a disorder of unknown etiology in which there is abnormal proliferation of non-Langerhans’ histiocytes. These, like Langerhans’ histiocytes, are probably a group of dendritic cells.29 It is characteristically a benign skin disorder of infants and young children which tends to undergo spontaneous regression. It is more common in children with neurofibromatosis type 1 (NF1) and can be the first sign of this disorder.30,31 The simultaneous presence of JXG and NF1 raises the risk of developing juvenile myelomonocytic leukemia 20–30 times,32 yet screening for leukemia in this group remains controversial.33 The skin lesions are occasionally accompanied by ocular involvement but ocular involvement can occur without cutaneous lesions. Visceral and bony involvement occurs less commonly than in LCH.34
Histopathology
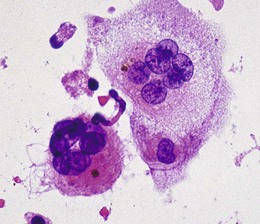
The JXG lesion consists of a mixture of lymphocytes, plasma cells, histiocytes, giant cells, and occasional eosinophils. The distinctive histologic feature is the presence of Touton giant cells, in which a central ring of nuclei encloses an area of eosinophilic cytoplasm surrounded by a foamy cytoplasm (Fig. 27.9). An important electron microscopic feature is the absence of Langerhans’ (Birbeck) granules in the histiocytes, distinguishing these lesions from LCH. Immunohistochemistry of JXG is positive for vimentin, CD68, and factor XIIIa immunostains and negative for S100 protein, helping to differentiate this condition from other histiocytic proliferations.29
Ocular involvement
JXG predominantly occurs in infancy and early childhood; in Zimmerman’s series34 85% of patients with ocular involvement were less than 1 year old, 64% less than 8 months. It occurs in neonates.35 Patients with ocular disease may occasionally present in adult life.36 Most have unilateral disease although a few cases with bilateral involvement have been reported.37 Cutaneous lesions are benign and often self-limited whilst ocular involvement can result in glaucoma and visual loss from optic nerve damage or amblyopia; however, since only 3 or 4 patients of every 1000 with cutaneous JXG develop ocular complications, routine eye screening of all patients is not justified. It could be limited to children with cutaneous involvement under 2 years of age, whose risk of ocular involvement is highest.38
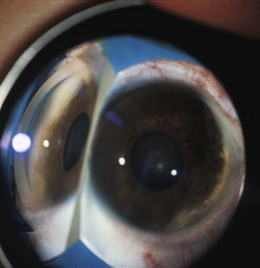
The iris is infiltrated in the majority of cases,34,39 the ciliary body,34,39 or rarely the choroid and retina39,40 may also be affected. Rarely, juvenile xanthogranuloma can masquerade as uveitis in childhood in the absence of skin lesions.39,41
Typically, a localized or diffuse yellow or fluffy-white iris lesion is evident in one eye of an infant (Fig. 27.10), often accompanied by hyphema (Fig. 27.11). Glaucoma, with corneal edema, photophobia, ocular enlargement, and circumcorneal flush are usually present. There may be uveitis and a xanthochromic flare. In some cases, iris heterochromia is the only presenting sign.