Chapter 41 Hereditary Vitreoretinal Degenerations
Hereditary vitreoretinal degeneration, also known as hereditary vitreoretinopathy, is classically characterized by early-onset cataracts, vitreous anomalies, coarse fibrils and membranes, and retinal detachment. Genetic and clinical advances in the last two decades have enabled a reassessment of the essential criteria that define this group of conditions. More recently, these conditions have been defined as the presence of congenital abnormalities of the vitreous, including severe degeneration or maldevelopment, early-onset progressive cataracts, and an increased predisposition to rhegmatogenous retinal detachment.1 Additional ocular and systemic features may be present, depending on the underlying cause.
In this chapter, we discuss five main types of vitreoretinopathies: (1) snowflake vitreoretinal degeneration; (2) X-linked juvenile retinoschisis; (3) the chromosome 5q vitreoretinopathies (e.g., Wagner syndrome); (4) chondrodysplasias with vitreoretinal degeneration (e.g., Stickler syndrome); and (5) enhanced S-cone syndrome (ESCS)/Goldmann–Favre vitreotapetoretinal degeneration. Several additional vitreoretinal degenerations are also summarized, including autosomal dominant neovascular inflammatory vitreoretinopathy (ADNIV) and autosomal dominant vitreoretinochoroidopathy (ADVIRC). We review the common features of vitreoretinal degenerations and highlight those features that can be used to distinguish them from each other clinically. Lattice degeneration and familial exudative vitreoretinopathy are not discussed in this chapter. Table 41.1 summarizes the main features of these disorders. Note that a given patient need not have all of the listed features. The pattern of clinical findings, rather than a specific feature, is the best strategy to make the diagnosis.
Snowflake vitreoretinal degeneration
General features
The term “snowflake vitreoretinal degeneration” (SVD) was originally coined by Hirose et al.2 in 1974, who described an American family of European extraction with early-onset cataracts, fibrillar vitreous degeneration, vascular sheathing, peripheral minute crystalline-like deposits, and retinal detachment. The condition received its name from the minute crystalline-like deposits that can be seen by contact lens biomicroscopy in some patients.2 The vitreous degeneration is fibrillar to a variable extent; the bands of condensed vitreous fibrils may obscure fundus details and peripheral condensations of vitreous on the retinal surface can be observed. Radial or circumferential lattice degeneration is not observed. The average spherical equivalent is –2.90 D, indicating moderate myopia. Other distinguishing features include a dysmorphic optic nerve head that appears flat and often without a cup. Peripapillary vascular sheathing and atrophy, as well as waxy pallor, may be present.
Clinical findings
Ocular features
There were 31 individuals in the Hirose family enrolled in the original study in 1974 (13 affected individuals, 14 unaffected individuals, and 4 unaffected spouses). The 13 subjects diagnosed with SVD ranged from 12 to 85 years of age, with clinical features such as early-onset cataract, fibrillar vitreous degeneration, vascular sheathing, and retinal detachment.2 The inheritance pattern was autosomal dominant, and no obligatory carriers of the snowflake trait were found to be normal. After about 20 years, Lee et al.3 restudied 6 of these 13 patients and identified additional clinical features, including corneal guttata (4 out of 5 patients) and optic nerve head dysplasia (the exact number of affected individuals was not recorded). Early-onset cataracts (5 out of 6), fibrillar vitreous degeneration (6 out of 6), and peripheral retinal abnormalities (5 out of 6), including minute crystalline-like deposits called snowflakes (4 out of 6), were common. Compared to other hereditary vitreoretinal degenerations, there was a relatively low rate of retinal detachment, occurring in 1 of the 6 examined family members.3 Orofacial features, early-onset hearing loss, and arthritis that are typical of Stickler syndrome were absent. Thus, the clinical findings were not typical of Stickler syndrome or chromosome 5q retinopathies, suggesting SVD may be a distinct form of vitreoretinal degeneration.
According to the dominant features present on fundoscopic examination, SVD has been classified into four stages: (1) extensive white with pressure; (2) snowflake degeneration; (3) sheathing of retinal vessels and fundus pigmentation; and (4) further pigmentation and disappearance of the peripheral retinal vessels. Hejtmancik et al. classified the clinical features of SVD into subgroups of congenital and progressive abnormalities.4 The congenital abnormalities include optic nerve head dysmorphism with fibrillar degeneration of the vitreous. Progressive ocular features include corneal guttae and peripheral retinal degeneration within which minute crystalline deposits, referred to as snowflakes, might be seen. These characteristics distinguish SVD from other vitreoretinal degenerations.
Using Hejtmancik’s genetic studies, it is evident that although the term “snowflake” has been used in reports of other families, they do not appear to be the same condition according to the clinical criteria and/or genetic evaluation. Moreover, the proportion of patients diagnosed with non-syndromic Stickler syndrome that ultimately share a common genetic basis with SVD patients is currently unknown, thus the real prevalence of SVD to date is difficult to estimate. So far, there is only one family reported in the literature with a case history similar to the classic description of SVD. This family was reported by Pollack and colleagues5,6 and showed an autosomal dominant vitreoretinal degeneration with minute crystalline-like dots, probably similar to the Hirose family. A distinguishing feature of this family, however was the appearance of neovascular tufts in the temporal periphery in 4 of the 9 affected members of the middle generation.6 Two other reports have described subjects with SVD. Robertson et al.7 reported familial clustering of granular deposits 100–200 µm in diameter in 10 patients from four families, which was described as snowflake degeneration of the retina in 1982. The deposits were evenly distributed about the circumference of the eye near the equatorial fundus. Smaller crystalline deposits were observed between the granular deposits. However, findings of vitreoretinal degeneration, such as early-onset cataract, severe vitreous degeneration, and retinal detachment, were not observed. The granular deposits are also not similar to those in patients with COL2A1 mutations causing Stickler syndrome.2,3 Chen and colleagues8 reported a family with vitreoretinal degeneration in 1986, but they were distinguished from the Hirose family by nyctalopia, poor visual acuity, annular scotomas, and attenuated retinal vessels. They may also have shown different-appearing deposits compared to the classical SVD description.2 Unfortunately, these two families have not been subject to genetic analysis.
Molecular genetics of SVD
Jiao et al.9 re-examined the family and localized the mutation to chromosome 2q36. Molecular genetic investigation excluded the known locations of genes causing vitreoretinal degeneration,3 and a novel gene location was subsequently identified. One of the authors (XD) studied the location of Kir7.1 (the protein product of KCNJ13) in the retina, and found that it is bound to the inner limiting membrane and the retinal pigment epithelium, which suggests that the mutation could affect development of the vitreous through alteration of Müller cell function.4 The mutation in KCNJ13 demonstrates that classic vitreoretinal degeneration can arise from gene mutations that are not structural components of the vitreous. Alteration in potassium transport provides a mechanism for the electrophysiological abnormalities seen in these patients, but further study is required for a precise explanation. The condition arises due to a mutation in KCNJ13 disrupting the selective transport of potassium through the channel.4 Thus, SVD can be clinically and genetically confirmed as a unique form of vitreoretinal degeneration.
Visual psychophysics
Kinetic perimetry shows peripheral defects, which are more pronounced in the superior field.10 Flicker perimetry reveals abnormalities undetected by kinetic perimetry and dark adaptation tests show elevated rod thresholds, except during the early stage of the disease.
Differential diagnosis
Stickler syndrome type I
This is the most common form of vitreoretinal degeneration. Mutations leading to haploinsufficiency of the collagen 2A1 (COL2A1) gene cause Stickler syndrome type I (STL1, MIM 108300). These patients have a vitreous degeneration characterized by a unique vitreous appearance vestigial vitreous gel occupying the immediate retrolental space and no discernible gel in the central vitreous cavity. The expression of syndromic features, including hearing loss, facial dysmorphism, and joint pain, exhibits variability both between and within families.11
Stickler syndrome type II
Mutations leading to haploinsufficiency of the collagen 11A1 (COL11A1) gene cause Stickler syndrome type II (STL2, MIM 604841). Unlike COL2A1 disease, these mutations lead to a fibrillar vitreous degeneration with limited and random fibrils throughout the vitreous cavity.12–14 In some cases, severe fibrillar degeneration of the vitreous can also be seen.
Marshall syndrome
Mutations altering intron–exon splicing of the COL11A1 gene lead to Marshall syndrome (MIM 154780), distinguished from SVD and Stickler syndrome by a more pronounced facial dysmorphism and lower frequency of retinal detachment.15,16 Membranous vitreous veils and radial lattice have also been noted in patients with Marshall syndrome.17
Wagner syndrome
Wagner syndrome (MIM 143200) is caused by noncoding mutations that are thought to affect the splicing of chondroitin sulfate proteoglycan-2 (CSPG2), the gene encoding versican.18,19 These mutations may lead to disease through abnormal ratios of versican isoforms. The distinguishing features of Wagner syndrome are pseudostrabismus, thickened and partially detached posterior hyaloids with an empty vitreous cavity, variable degeneration of the retina and choroid, and the absence of systemic manifestations.20 The absence of nyctalopia, posterior chorioretinal atrophy, and tractional retinal detachment distinguishes Wagner syndrome from other chromosome 5q vitreoretinopathies such as Jansen syndrome and erosive vitreoretinopathy (ERVR).
Goldmann–Favre vitreotapetoretinal degeneration
Patients with Goldmann–Favre vitreotapetoretinal degeneration have nonrecordable ERGs and central and/or peripheral retinoschisis. This is in contrast to patients with snowflake degeneration, who have fairly good ERG responses and absence of central or peripheral retinoschisis.21
Management
Common issues in the management of hereditary vitreoretinal degenerations are described below:
1. Cataract surgery is difficult in these patients due to lack of vitreous support during surgery, and should be performed by experienced surgeons with specific experience with vitrectomized eyes that behave similarly. Microinvasive 23- or 25-gauge infusion cannulae via the pars plana are beneficial for maintaining the intraocular pressure.
2. Glaucoma can occur, usually after cataract surgery, and should be monitored and treated using established methods.
3. Prophylactic cryopexy is performed by some groups, while peripheral laser retinopexy is favored by others. Although a randomized trial has yet to be published comparing the two methods, a recent report using cryopexy is the most comprehensive study to date.22 Ang et al. found that, in patients with type I Stickler syndrome, the prevalence of RD was significantly less in bilateral 360° prophylactic cryotherapy than that in untreated patients, suggesting that prophylactic cryotherapy may be substantially beneficial.22
4. Retinal detachments are also common and should be managed using standard vitrectomy-based approaches, including vitrectomy, artificial posterior vitreous detachment, and release of peripheral traction, perfluorocarbon-mediated retinal reattachment, scleral buckle, laser retinopexy, and tamponade with gas or silicone oil.
The chromosome 5Q retinopathies: wagner syndrome, jansen syndrome, erosive vitreoretinopathy, and related conditions
General features
Wagner syndrome is characterized by an optically empty vitreous with avascular vitreous strands and veils, moderate myopia, presenile cataracts, and retinal degeneration with atrophy.23 Stickler syndrome is also associated with craniofacial abnormalities and a progressive arthropathy.24 Wagner syndrome and Stickler syndrome were once incorrectly considered as one entity: the Wagner–Stickler syndrome.
Wagner syndrome is an autosomal dominant genetic disorder first mapped to chromosome 5q13–14 in 1995. A mutation in the chondroitin sulfate proteoglycan 2 gene (CSPG2), now named VCAN, encoding for the versican protein, was found in 2005.20,25,26 VCAN is the only gene currently associated with Wagner syndrome and ERVR.19,27 Autosomal dominant Stickler syndrome is an inherited progressive disorder of the collagen connective tissues and is associated with the mutation of extracellular matrix collagen genes, such as COL2A1, COL11A, COL11A2, and COL9A1.24,28–30
Jansen syndrome was described as vitreoretinal and lenticular degeneration associated with retinal detachments in the absence of nonocular findings. However, the disease gene for the original Jansen family was demonstrated to be linked to the same region of chromosome 5q1431 where genes for Wagner syndrome and ERVR were located.25
ERVR also displays an autosomal dominant inheritance pattern and shares some clinical features with Wagner syndrome. The critical region of the genetic defect underlying ERVR was found to overlap with the critical region for Wagner syndrome, 5q13-q14.19
Clinical findings
Ocular features
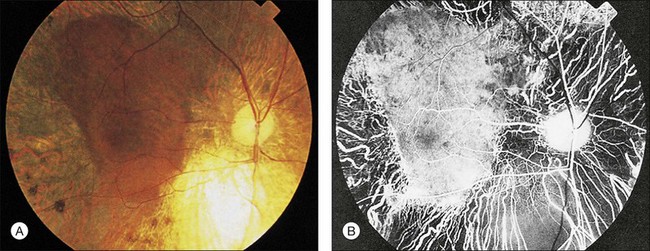
Wagner syndrome is characterized by an optically empty vitreous with equatorial avascular vitreous veils. Additional features include its early-onset, moderate myopia, and typical dot-like cortical cataracts, foveal ectopia, abnormal retinal vessels (inverted papilla), perivascular pigmentation and sheathing, retinal thinning, as well as slowly progressive chorioretinal atrophy. Patients with Wagner syndrome have pseudostrabismus from congenital temporal displacement of the fovea. They also have nyctalopia early in life and final dark adaptation thresholds are elevated in some patients.32 Most patients under the age of 20 have normal vision; however, cataract, retinal detachment, optic atrophy, and chorioretinal atrophy may be progressive and cause visual loss with advancing age20,23,25–27 (Fig. 41.1). In addition to the clinical features of Wagner syndrome, ERVR reveals progressive nyctalopia and visual field constriction. A conspicuous loss of retinal pigment epithelium and choriocapillaries is observed by fluorescein angiography.19 The vitreous findings are marked syneresis with prominent membranes. Rhegmatogenous retinal detachment is usually described as a feature of ERVR, and is less frequent in the original description of Wagner syndrome.33
Visual psychophysics
Nyctalopia can be present early in life in some patients. Vision is usually normal as the pathologic process initially involves the retinal periphery, but severe loss of vision will occur in patients when diffuse cone–rod loss ensues as there is progressive chorioretinal atrophy.1,25
Electrophysiology
Both the ERG and dark adaptation of patients with the chromosome 5q retinopathies appear to be normal early in life but become progressively abnormal throughout the patient’s life. The rod and cone systems are affected to varying degrees but in a family-specific manner. While both a-wave and b-wave amplitudes are reduced, b-wave amplitudes are generally better preserved. Visual field findings can be variable and may include diffuse peripheral loss or partial/complete midperipheral ring scotomas as the chorioretinal atrophy progresses.34
Differential diagnosis
Autosomal dominant vitreoretinopathies
Snowflake vitreoretinal degeneration
SVD is a progressive hereditary eye disorder caused by mutations in KCNJ13. Diagnostic features of SVD consist of fibrillar vitreous degeneration, early-onset cataract, minute crystalline deposits in the neurosensory retina, and retinal detachment.4 However, membranous degeneration of the vitreous with avascular strands and veils is not observed in SVD. Retinal defects typically start in the superficial retinal layers and retinal detachment is uncommon.4,27
Stickler syndrome
Stickler syndrome is genetically distinguished from Wagner syndrome and other chromosome 5q retinopathies. Type I Stickler syndrome is due to COL2A1 mutation and is associated with retrolental membranous vitreous, while type II Stickler syndrome is due to mutations in COL11A1 and is associated with a fibrillar or beaded vitreous phenotype. Both type I and II Stickler syndrome have ocular and systemic manifestations, while type III Stickler syndrome, associated with COL11A2 mutations, has systemic manifestations only. Most, but not all, patients with Stickler syndrome have congenital, nonprogressive, and high-degree myopia. The cataracts may be congenital and nonprogressive, and have an unusual characteristic curved cortical distribution.35 Retinal detachment is much more common in Stickler syndrome (50%) than in chromosome 5q vitreoretinopathies (15%). Systemic abnormalities are present in Stickler syndrome, such as midface hypoplasia, midline cleft of the palate, bifid uvula, sensorineural hearing loss, and skeletal abnormalities.24,27–29 Abnormal dark adaptation associated with alterations in the ERG that is common in chromosome 5q retinopathies has not been described in Stickler syndrome.1,27
Autosomal dominant vitreoretinochoroidopathy
ADVIRC is caused by mutations in VMD2 and also has characteristic retinal and vitreous findings, in particular a peripheral retinal circumferential hyperpigmented band, vitreous fibrillar condensation, punctate white opacities in the retina, breakdown of the blood–retinal barrier, and retinal neovascularization.36
Autosomal recessive vitreoretinopathies
Goldmann–Favre syndrome (GFS) and enhanced S-cone syndrome
GFS and ESCS share common mutations in the NR2E3 gene and are usually associated with night blindness and visual field abnormalities. ERG typically reveals a severe reduction in rod function and a relatively enhanced function of the short-wavelength-sensitive cones.27,37 GFS manifests with progressive vitreous changes, hemeralopia, chorioretinal atrophy, and pigmentary retinal degeneration, later resulting in marked visual field loss, retinoschisis in the periphery and/or macula, presenile cataract, and hyperopia rather than myopia.27,38,39 ESCS lacks the typical marked vitreous changes of GFS.
Treatments
Refractive error is corrected by spectacles or contact lenses. Cataract is managed by the phacoemulsification and implantation of an intraocular lens. Retinal breaks without retinal detachment are treated with laser photocoagulation. Vitreoretinal surgery is needed for retinal detachment, vitreoretinal traction involving the macula, or epiretinal membranes involving the macula. Prophylactic cryotherapy has been recently reported to reduce the risk of retinal detachment markedly.1
Chondrodysplasias associated with vitreoretinal degeneration: the stickler syndromes, marshall syndrome, kniest dysplasia, knobloch syndrome, and weissenbacher–zweymuller syndrome
General features
Chondrodysplasias refer to a group of hereditary and systematic disorders that affect skeletal development and growth. These conditions may also feature ocular, central nervous system, or renal abnormalities. The genes involved in these syndromes affect types II, III, V, X, or XI, collagen molecules that are found mainly in cartilage and vitreous and are essential for the normal development of bones and other connective tissue, thus accounting for the symptoms observed clinically. Antenatal diagnosis, including fetal magnetic resonance imaging, computed tomography, ultrasonography, and genetic testing of fetal DNA obtained from aminocentesis or chorionic villus sampling, is important for the proper management of children and counseling of parents.41–43
Stickler syndrome, also known as hereditary progressive arthro-ophthalmopathy, is considered to be the most common chondrodysplasia associated with vitreoretinal degeneration. Types I, II, and III are the three subgroups of Stickler syndrome classified by genetic heterogeneity. Types I and II Stickler syndrome, respectively caused by mutations in the COL2A1 gene encoding type II collagen44 and in the COL11A1 gene encoding type XI collagen,45 can be differentiated successfully by vitreous phenotypes. Mutations in COL2A1 usually result in a congenital membranous vitreous anomaly, while mutations in COL11A1 result in an irregular and beaded vitreous.46,47 Recently, a new subgroup of COL2A1 mutations was found that lead to a hypoplastic vitreous which is either optically empty or contains sparse irregular lamellae.13 Due to the differential splicing, COL2A1 gene transcription products can be divided into two types: collagen IIA with an exon2-encoded length of 69 (rich in amino acid homocysteine) and collagen IIB without the exon2-encoded peptide. Whereas both collagen IIA and IIB loss results in systemic connective tissues disease, collagen IIA loss also leads to ocular abnormalities. It is because collagen IIA primarily exists in the vitreous.48,49 Recent studies have indicated that mutations in any collagen IX genes, like COL9A1, COL9A2, or COL9A3, can cause autosomal recessive Stickler syndrome.50–52 Thus far, the diagnosis of Stickler syndrome has been based on clinical manifestations without consensus on the minimal clinical diagnostic criteria. The anomalous formation of the vitreous gel structure and its corresponding characteristic abnormalities are essential for the diagnosis of types I and II Stickler syndrome. In additional to these clinical manifestations, a detailed family history should be obtained and the diagnosis can be confirmed by genetic analysis. Recently, the COL2A1 mutation in peripheral white blood cells has been used to identify patients with type I Stickler syndrome.51
Marshall syndrome, an autosomal dominant chondrodysplasia, is caused by a splicing mutation of 54-bp exons in the c-terminal region of the COL11A1 gene which is located on the short arm of chromosome 1. Although Marshall syndrome and Stickler syndrome are now considered two distinct diseases, a case of a patient with overlapping phenotypes has been described.53
Knobloch syndrome is a rare and clinically heterogeneous autosomal recessive disorder. Collagen XVIII, which is a basement membrane proteoglycan, distributes in multiple organs of the body and plays an important role in the function and development of the eye, kidney, and nervous system. In most patients with this symptom, null mutations in the COL18A1 gene mapped to the long arm of chromosome 21 are supposed to induce the changes in collagen XVIII.54 There are also case reports of Knobloch syndrome without COL18A1 gene mutations, and thus immunofluorescent histochemistry of skin biopsy samples has proved to be a useful preliminary and complementary test for the diagnosis of Knobloch syndrome.55
Weissenbacher–Zweymuller syndrome, also called Pierre Robin syndrome with fetal chondrodysplasia, is an autosomal recessive disorder characterized by a single-base mutation in the COL11A2 gene, which substitutes glutamate for glycine.56 In the past, it was often misdiagnosed as Stickler syndrome. Recently, investigators have differentiated these two syndromes successfully by prenatal ultrasonography.57
Clinical findings
Extraocular features
Stickler syndrome features a highly variable systemic phenotype, including conductive and sensorineural hearing loss, immunoglobulin deficiency,20 cleft palate, mid facial underdevelopment, mild spondyloepiphyseal dysplasia, and precocious arthritis58 (Fig. 41.2).

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


