Chapter 43 Hereditary Choroidal Diseases
 For additional online content visit http://www.expertconsult.com
For additional online content visit http://www.expertconsult.com
Introduction
Nonetheless, the hereditary choroidal dystrophies can be classified in the following manner: (1) choroidal atrophy phenotypes, which can be further subdivided into: (a) central areolar choroidal dystrophy (CACD); (b) peripapillary choroidal dystrophy; and (c) diffuse choroidal dystrophy; (2) gyrate atrophy of the choroid and retina; and (3) choroideremia (CHM). While each of the choroidal dystrophies has characteristic fundus features, in certain instances at advanced stages of disease an overlap in fundus appearance may be observed (Table 43.1).
Table 43.1 Brief outline of chapter layout
Choroidal atrophy phenotypes
According to Sorsby,1 this group of disorders can be subdivided into three clinical phenotypes based on their geographical distribution. They include: central areolar, peripapillary, and more diffuse or generalized choroidal dystrophy. All can be inherited as either autosomal dominant or autosomal recessive traits.
Central areolar choroidal dystrophy
CACD was first described by Nettleship in 1884. It is inherited primarily as an autosomal dominant trait,2,3 although autosomal recessive cases have been occasionally reported.4,5 Yanagihashi and colleagues6 identified a novel mutation in the peripherin/RDS (retinal degeneration slow) gene in a Japanese family with an autosomal dominant form of CACD.
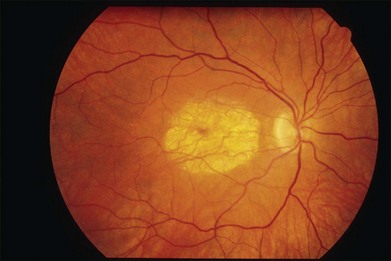
The initial symptoms of diminished central vision generally begin in the latter part of the second to the early part of the fourth decade. Characteristic bilateral macular lesions are solitary with well-defined margins, and circular or ovoid in shape (Fig. 43.1). Although they may increase in size and become irregular in shape, they do not involve the peripapillary region or extend beyond the vascular arcades.
Peripapillary choroidal dystrophy
The peripapillary form of choroidal dystrophy is usually inherited as an autosomal recessive trait,7 although in some instances autosomal dominant transmission may be encountered.
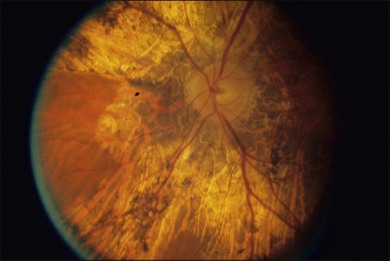
The fundus findings in this form of choroidal dystrophy initially include changes seen in the RPE and later an ophthalmoscopically apparent loss of RPE and choroidal tissue. The important distinction between CACD and the peripapillary phenotype is their location. The peripapillary form begins in the region surrounding the optic disc and slowly enlarges, in finger-like projections, nasally, temporally, and into the macula, eventually occupying the entire posterior pole (Fig. 43.2). In some instances the peripapillary form can progress to a phenotype similar to the diffuse form.
Visual field and dark-adapted final thresholds testing indicate that peripheral to the fundoscopically involved area, retinal function is either normal or mildly impaired. The full-field ERG is either normal or only slightly reduced, reflecting the extent of the disease.7,8
The differential diagnosis includes peripapillary pigment epithelial dystrophy in which there are well-defined areas of RPE loss, with direct visualization of the underlying choroidal vasculature. Fluorescein angiography shows an intact choriocapillaris that differentiates it from those with choriocapillaris loss.7 Another disorder that mimics peripapillary choroidal dystrophy is serpiginous choroiditis, which usually begins in the peripapillary region and then extends into the retina in pseudopod-like extensions, sometimes involving the macula.
Diffuse choroidal dystrophy
This diffuse disorder of the RPE and choriocapillaris is most often inherited as an autosomal dominant trait;9 however, autosomal recessive transmission may occur. The onset of symptoms occurs most often in the fourth and fifth decade and is usually manifested by poor central vision, impairment of night vision, or both.
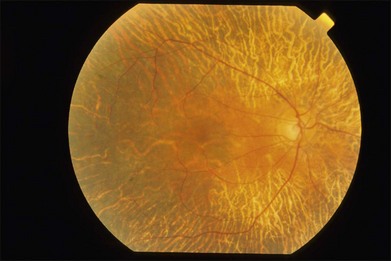
The early fundus changes include retinal pigment mottling and hypopigmentation. The disease may initially show a predilection for the posterior pole of the retina before progressing to a more diffuse phenotype. Later there is diffuse atrophy of both the RPE and choriocapillaris while the larger choroid vessels appear sclerotic as yellowish-white bands. Both the posterior pole and the periphery are involved to varying degrees. Even with diffuse involvement in the more advanced stages, the retinal vessels usually remain normal10 (Fig. 43.3). In the end stages, diffuse choroidal dystrophy cannot be easily differentiated from other diffuse chorioretinal diseases such as thioridazine (Mellaril) retinal toxicity, advanced stages of both pattern dystrophy and Stargardt disease, in addition to the advanced retinopathy seen in the Kearns–Sayre syndrome.
Psychophysical and electrophysiological studies reflect the diffuse involvement. Visual fields show a concentric peripheral constriction, while ERG recordings are either subnormal10 or undetectable.7 Fluorescein angiography shows a loss of the choriocapillaris and visualization of the larger choroidal vessels beneath atrophic-appearing RPE.7,11 A few scattered areas show a patchy choroidal flush pattern indicative of some remnants of the choriocapillaris.
Gyrate atrophy of the choroid and retina
The first case of this disease was described in 1888 by Jacobsohn12 as an example of “atypical retinitis pigmentosa”; however, Cutler13 in 1895 and Fuchs14 in 1896 were the first to recognize the disease as a distinct clinical entity. Gyrate atrophy is a rare choroidal disease with a prevalence of about 1 in 50 000 in Finland.15 It is inherited as an autosomal recessive trait, although dominant pedigrees have also been reported.10 The biochemical abnormalities observed in this disorder were initially described by Simell and Takki16 in 1973; these included a deficiency of the enzyme ornithine-delta-aminotransferase (OAT), which results in an increase in the plasma ornithine concentration (10–15 times the normal levels). The enzyme OAT is a mitochondrial-encoded enzyme with pyridoxal phosphate (vitamin B6) enzyme as a cofactor that catalyzes the interconversion of ornithine, glutamate, and proline. This results in systemic biochemical abnormalities, including hyperornithinemia, and reductions in plasma lysine, glutamine, glutamate, and creatine.17–19 Either an absence or a marked reduction of OAT in cultured skin fibroblasts and in lymphocytes has been observed.20 A number of different mutations have been identified within the OAT gene on chromosome 10.21–23 Kellner and colleagues24 previously described a gyrate atrophy-like phenotype in 6 male patients, 3 of whom were patients of the same family, with normal serum ornithine levels.
The onset of visual symptoms, including poor night vision and constricted peripheral vision, usually begins in the second and third decades. Since both structural and visual functional changes spread from more peripheral to a central location, loss of visual acuity is a later complaint in the disease. Myopia and posterior subcapsular cataracts are frequently observed and vitreous opacities may also be present.15
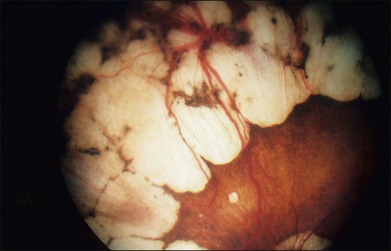
The fundus changes begin in the midperipheral and peripheral retina with a thinning and atrophic appearance of the RPE in which the underlying choroidal vessels may appear either normal or sclerotic. These areas are typically scalloped in shape and are initially separate but tend to become confluent as they slowly progress both centrally and peripherally (Fig. 43.4). Progression of the disease leads to pigment clumping, RPE and choriocapillaris atrophy, and eventual total atrophy of the choroid exposing the white sclera. In the late stages, an annular ring of choroidal atrophy may be seen from the periphery to the posterior pole, usually sparing the macula. The retinal vessels may appear normal initially or attenuated in later stages of the disease when the optic nerve may appear pale.7,10
There are reports on the presence of cystoid macular edema in patients with gyrate atrophy.25–27 A study by Vasconcelos-Santos et al.27 showed a short-term therapeutic effect with the use of a 4-mg intravitreal triamcinolone acetonide injection for gyrate atrophy-related macular edema. After drug clearance, the edema recurred, with return of visual acuity to the pretreatment level.
Visual function varies considerably from case to case and seems to be related to the extent of fundus involvement. Visual field testing shows a concentric peripheral constriction of the visual field as the most often observed abnormality. However, an annular ring and paracentral scotomas may develop as the disease progresses. Eventually, if the fovea becomes involved, a central scotoma will be seen.7,28
Early in the disease, dark adaptation testing shows only mild threshold elevations while significant elevation of the final rod thresholds is eventually noted in most patients. In the early stage, full-field ERG recordings may show only a mild abnormality in rod and cone amplitudes, while, as the disease progress, the ERG responses deteriorate and may eventually become undetectable. The rod responses are affected more severely in the early stages, but later both cone as well as rod function is severely impaired.29,30 The electro-oculogram (EOG) is normal or only mildly reduced at very early stages. The EOG light peak to dark trough ratio becomes markedly reduced in the later stages. Electromyograms are often abnormal, although only a few patients complain of mild muscle weakness. Muscle biopsy shows atrophic type 2 muscle fibers with tubular aggregates visible on electron microscopy.31,32 Both electrocardiographic and electroencephalographic abnormalities may be observed in some patients.33,34 Histopathologic studies show early changes in RPE cells, with subsequent loss of photoreceptors and choriocapillaris, suggesting that these latter changes may be secondary to the loss of RPE cell integrity.35 A report of a histologic study in the ornithine-deficient mouse model of gyrate atrophy36 is available online.
A histopathologic study in the ornithine-deficient mouse model of gyrate atrophy showed earliest changes in the RPE cells in the form of sporadic degeneration of scattered cells. By 6 months, there were more diffuse abnormalities of the RPE with accumulation of large phagosomes and crystalloid inclusions. Although morphologically normal at 2 months, the photoreceptor outer segments became highly disorganized and shortened to 60% of mouse control lengths by 10 months. Additionally, there was a cumulative loss of the photoreceptor cells, which reached 33% by 10 months.36
An arginine-restricted diet has been pursued as a form of therapy in patients with gyrate atrophy of the choroid and retina.17,37–39 Since ornithine is produced from other amino acids, mainly arginine, some investigators advocate that patients be restricted to a rigid low-protein diet, including near-total elimination of arginine with supplementation of essential amino acids. Orally administered pyridoxal phosphate (vitamin B6), can result in a reduction in plasma ornithine levels in some patients, while others are nonresponsive to B6.29 Overall, ERG responses are better maintained by B6-responsive patients compared to those who are nonresponders.29,37 While Kaiser-Kupfer et al.38 concluded that a dietary approach to reducing plasma ornithine was effective, this was not similarly observed to occur in patients with gyrate atrophy in studies by Vannas-Sulonen et al.39 or Berson et al.37
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


