Gyrate Atrophy
Steve A. Arshinoff
Kevin Leung
Yi Ning J. Strube
HISTORY
Gyrate atrophy (GA) (MIM 258870) of the choroid and retina is a rare autosomal recessive disorder characterized by progressive, metabolic, retinal, and choroidal degeneration due to deficiency of the pyridoxal phosphate (PLP)–dependent, nuclear-encoded, mitochondrial matrix enzyme ornithine delta(δ)-aminotransferase (OAT; L-ornithine:2-oxoacid aminotransferase; EC 2.6.1.13), which has been mapped to chromosome 10q26.1,2 GA was initially described as an atypical type of retinitis pigmentosa by Jacobsohn in 18883 and by Cutler in 1895.4 The disease was given its current name by Fuchs in 1896.5 The extreme rarity of this disorder is evident from the fact that when Usher6 reviewed all previously known cases in 1935 and correctly concluded that the inheritance pattern was autosomal recessive, he could find reports of only 26 cases. When Kurstjens7 did his extensive review in 1965 and characterized many of the typical ophthalmic findings, he could find only 44 cases in the world literature. A careful review at the present time still reveals just over 150 biochemically documented cases, about one-third of which are from Finland and only seven (less than 5%) of which have been responsive to therapy with vitamin B6 dietary supplementation.8
Simell and Takki’s discovery of associated hyperornithinemia, the primary biochemical manifestation of OAT deficiency, in 1973 caused an explosion of interest in this disease, making it definable metabolically as well as ophthalmologically.9 Since then, GA has been transformed from an obscure curiosity to the prototype of potentially treatable metabolic retinal degenerations. It was also the first primary retinal degeneration to be recognized as an inborn error of metabolism, as defined by Garrod in 1908.10 Readers interested in metabolic details of GA beyond this chapter are referred to the chapter by Valle and Simell in the eighth edition of The Metabolic and Molecular Bases of Inherited Disease.8
DIAGNOSIS
The diagnosis of GA can easily be made with certainty if the five most prominent features can be recalled.11
Typical gyrate retinal and choroidal lesions (Fig. 1): The appearance of confluent arcuate equatorial full-thickness lesions of the choroid and retina, sparing some of the larger choroidal vessels and separated from one another by thin margins of pigment, is characteristic of GA. The lesions extend without interruption to the ora serrata, where a dense aggregation of pigment can be seen, but they invade the circummacular vascular arcade only in advanced cases. Peripapillary gyrate lesions are common, but foveal lesions are rare until late in the course of the disease.
Early cataract: Posterior subcapsular lens changes usually begin in the late teens, and fully developed posterior subcapsular cataracts with diffuse cortical opacities are almost invariably present by age 30.
High myopia with marked astigmatism: Myopia of -6 to -10 diopters (D) or more, accompanied by 2 D or more of astigmatism is present in about 90% of cases so far reported. The occasional case may be only mildly myopic and astigmatic.
Hyperornithinemia: Extreme hyperornithinemia is universal in this disease. Its absence necessitates a search for another diagnosis. All body fluids measured to date (whole blood, plasma, cerebrospinal fluid, aqueous humor, and urine) have been found to contain 10 to 20 times the normal levels of ornithine.9,11
Autosomal recessive inheritance pattern: To date, all patients with the oculometabolic disease have family trees and laboratory genetic findings consistent with autosomal recessive inheritance. It is possible to diagnose heterozygotes and to confirm the inheritance pattern in a family with a single affected member (see below).12
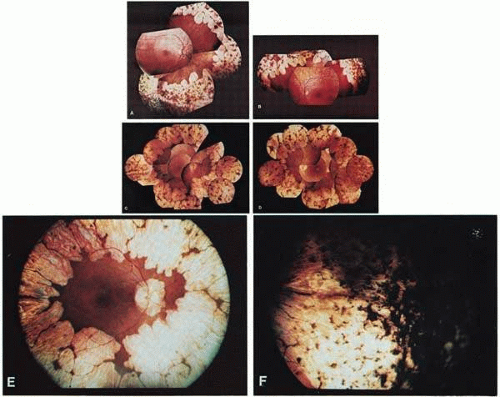 Fig. 1. Representative fundus photographs of patients with gyrate atrophy (GA). A and B. The right and left fundi, respectively, of a 9-year-old white female patient with relatively early lesions. There is total sparing of the retina within the circummacular vascular arcade and a lack of pigment between the gyrate lesions (corrected visual acuity, 20/30 RE, 20/25 LE). C and D. The appearance of the right and left fundi, respectively, of a 16-year-old white male patient. The lesions extend considerably farther posteriorly than in the younger patient in A and B. Also, in the left eye, there is increased pigmentation between the lesions, as well as a foveal lesion and peripapillary lesions (corrected visual acuity, 20/100 RE, 20/80 LE). E. The posterior pole in the right eye of a 31-year-old white female patient with advanced GA. The gyrate lesions extend within the circummacular vascular arcade; a large peripapillary lesion is present. F. The far periphery of the eye shown in E. There is extensive accumulation of pigment, typical of advanced GA. |
OPHTHALMIC FINDINGS
A detailed ophthalmic examination of a patient with GA usually reveals the following findings:
Visual acuity: Acuity in most cases is normal with correction, up until about age 10. There appears to be a gradual decline in visual acuity thereafter, unrelated to the development of cataracts. The slowly progressive irreversible loss of vision from retinal degeneration often leads to blindness, usually by the fourth or fifth decade of life.13 The appearance of a large peripapillary gyrate lesion is often accompanied by a decline in central vision. Rarely, a patient may develop a foveal gyrate lesion during the early stages of the disease, but these lesions are often present in advanced cases. Occasionally, acuity is inexplicably decreased in one eye, when compared with its fellow, in the same patient. Variation in the severity of visual loss among patients from different families is considerable and may be due to the tremendous variety in the specific mutations in the GA gene found in different families (see metabolic discussion below; e.g., pyridoxine-responsive mutations tend to be less severe).
Pupils: Pupillary appearance and reactivity to light and near are generally normal. Patients with marked asymmetry in acuity, visual fields, and other parameters may have an afferent pupillary defect in the worse eye.
Motility: Extraocular muscle balance is usually normal, except for a moderate exophoria not unlike that seen in other myopes. Patients with significantly worse vision in one eye may have manifest exotropia.
Stereopsis: Stereopsis is normal as long as the visual acuity remains symmetric. A decrease in stereopsis may be an early sign of developing exotropia and decline in the visual acuity of one eye.
Color vision: Decreased color discrimination occurs when there is a drop in visual acuity on the basis of retinal lesions, or with the development of cataracts. Otherwise, there are no remarkable changes in color vision, as the central color sensitive cones appear to be preserved until late in the disease.14
Visual fields: Visual fields coincide fairly well with the remaining area of healthy-appearing retina (i.e., about 15 to 50 degrees), with an enlarged blind spot corresponding to the peripapillary lesion, if present. Using fundus photoperimetry, Enoch and associates15 found that the limit of the functional visual field typically conforms to the observed gyrate lesions with a sharp drop-off in visual function at the border of the lesions. However, the functional visual field may occasionally be considerably smaller than the central area of the retina that appears to be healthy on ophthalmoscopy, and this is often associated with a large peripapillary lesion.11
Lens: Posterior subcapsular cataracts develop in the late teens or early twenties and progress to include extensive cortical changes primarily involving opacification of the lens along the suture lines, extending toward the equator of the lens.16 Anterior subcapsular fibrous plaques have been reported.17
Ciliary body: Abnormally short and scant ciliary processes have been reported from cycloscopic examination of these patients.18
Vitreous: The vitreous is typically syneretic and frequently contains clusters of cloudy fibrils. Recurrent vitreous hemorrhages during adolescence have been noted in a few patients.19,20,21
Fundus: Gyrate appearance is typical (see Diagnosis section). Takki and associates have described the presence of crystals in the demarcation lines of the peripheral gyrate lesions in some patients.9,12,22 Choroidal large vessels may be seen where the retina and pigment epithelium have atrophied. At the macula, a granular appearance may be observed before atrophic changes become evident. A zone of pigmentary change may separate normal and atrophic areas. These areas may represent abnormally functioning retina and pigment epithelium prior to the stage of visible atrophy.22,23 Bakker and colleagues24 described subretinal neovascularization and foveal disciform hemorrhages in a case of GA. Marano and colleagues25,26 described GA associated with low-activity subfoveal choroidal neovascularization (CNV) development, possibly a result of the high myopia present.
Intraocular pressure: Alteration in intraocular pressure has not been described in the literature as an associated finding in GA. Since 1977, however, the author (SAA) has been following a patient who developed typical pigmentary glaucoma 5 years after bilateral intracapsular cataract extraction, with dense bands of presumed retinal pigment epithelial (RPE) pigment in the trabecular meshwork bilaterally. This patient has been successfully managed with filtration surgery and topical medications bilaterally. Subsequent patients have had their cataracts managed with extracapsular cataract extraction or phacoemulsification and have not developed glaucoma, possibly because of preservation of the posterior capsule as a barrier to pigment migration (Steve A. Arshinoff, unpublished data, 1977–2003).
Fluorescein angiography: There is mild leakage of fluorescein at the margin of the healthy-appearing retina, where it abuts the gyrate lesions (Fig. 2). The earliest detectable changes include pigment epithelial transmission defects, which eventually progress into patches of atrophy with drop-out of the choriocapillaris in the involved area. The edges of such patches stain by leakage, indicating functioning choriocapillaris in the surrounding tissue.27 The ophthalmoscopically normal areas of retina appear normal on fluorescein angiography.
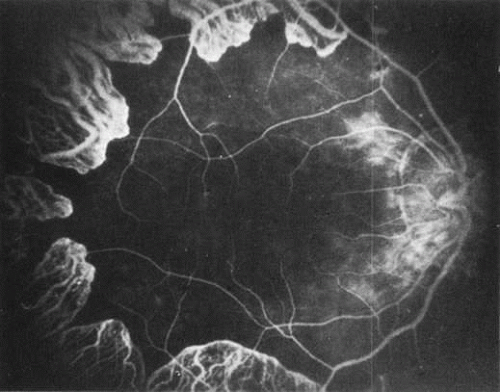
Fig. 2. A fluorescein angiogram shows slight leakage from the margins of the choriocapillaris neighboring the atrophic areas. The posterior pole appears surprisingly normal.
Dark adaptation: The dark adaptation curve is usually monophasic, delayed, and elevated above normal by about 3 log units.
Electroretinography: The full-field electroretinogram (ERG) has been shown to be reduced in the early stages of GA, and no normal ERG recordings have been reported.28,29 Both rod- and cone-mediated a- and b-wave amplitudes are usually severely reduced (less than 10 μV) or even isoelectric, indicating that rod and cone systems are affected jointly, although occasionally a patient’s ERG may be reduced by only 50% to 75% (i.e., recordable at 100 μV to 300 μV). Direct current ERG findings indicate that the c wave can be recorded only in the very early stages of the disease. The disappearance of the c wave prior to the a and b waves in GA suggests that the ocular disease is primarily RPE in origin.30 Some patients have a delayed implicit time on 30 Hz flicker testing. Computer averaging with narrow-band filtering has made it possible to detect ERG responses under 1 μV, allowing objective monitoring of the progress of even severely affected subjects.31
Electro-oculography: The ratio of the light peak to the dark trough is severely reduced, parallel to the reduction in the ERG.
Visual evoked response: The visual evoked response appears to vary as a direct function of visual acuity.
Conjunctival pathology: Three conjunctival biopsies of patients with GA have shown changes in the epithelial cells and in the stromal fibroblasts of the conjunctiva. The presence of osmiophilic particles (hypothesized to be lipidic or fatty acid drops), hypertrophy of the Golgi apparatus with rupture of intracellular membranes, and accumulation of lysosomes have been detected on electron microscopy.32
Whole-globe pathologic examinations: A postmortem histopathologic examination of whole globes from a 98-year-old woman with a well-documented (confirmed with fibroblast cultures and hyperornithinemia), atypically mild case of vitamin B6–responsive GA, who had retained 20/50 vision up until death, was reported by Wilson and associates.33 Distinctive, circumscribed patches of RPE atrophy were observed only at the periphery. The outer retina, RPE, choriocapillaris, and most of the choroidal vessels were absent from these patches. Where the RPE terminated, the photoreceptor cells directly abutted Bruch’s membrane. On electron microscopy, the mitochondria in the corneal endothelium, nonpigmented ciliary epithelium, smooth muscle of the iris, and ciliary body were enlarged and had disrupted cristae and an electronlucent matrix. Similar but less severe mitochondrial abnormalities were found in the photoreceptors. No mitochondrial abnormalities were found in the RPE.
SYSTEMIC CLINICAL AND PATHOLOGIC FINDINGS
Although ocular involvement is the most clinically significant manifestation of GA, other organs may be affected. On systemic examination, ornithine levels about 10 to 20 times normal values (fasting morning plasma ornithine = 400 to 1400 μM [normal = 40 to 120 μM, mean = 60 to 80 μM]) were observed.8 Hyperornithinemia is an essential component of GA and was the finding that proved GA to be an inborn error of metabolism, with the eye as the most apparent target organ of the metabolic derangement. McCulloch and Marliss34 demonstrated that ornithine was being released from many organ systems and suggested multisystem involvement. The following organs and tissues have so far been found to be affected in GA:
Brain: Takki22 first reported that abnormal electroencephalographic (EEG) recordings and borderline low intellectual function were common in GA. McCulloch and coworkers11,34 confirmed these findings and stated that they do not necessarily coexist in the same patient. Kaiser-Kupfer and associates35 also reported EEG abnormalities in GA. Using brain magnetic resonance imaging (MRI), Valtonen and colleagues36 found degenerative lesions in the white matter of 50% of 23 GA patients studied and premature atrophic changes in 70%, with a striking increase in the number of Virchow spaces. In agreement with the above-mentioned studies,11,22,32,35 EEG recordings revealed abnormal slow background activity, focal lesions, or high-amplitude B rhythms (greater than 50 mV) in 58% of 33 GA patients tested. There was no correlation between EEG and MRI results or the age or sex of the patients. The MRI and EEG results did not differ between untreated and creatine (Cr)-supplemented GA patients. The authors concluded that early degenerative and atrophic brain changes and abnormal EEG findings are additional clinical features of GA. Nanto-Salonen and colleagues37 analyzed the proton magnetic resonance (MR) spectra of the basal ganglia in 20 GA patients. They found reduced brain Cr stores in GA, which was partially corrected by low-dose Cr supplementation and an arginine-restricted diet. They concluded that these results support the hypothesis that hyperornithinemia in GA results in chronic Cr depletion and decreased phospho-Cr (PCr) stores in the retina, central nervous system, and muscle, and may contribute to the observed pathology in those organs.
Peripheral nervous system: Peltola and colleagues38 used neurography, quantitative sensory threshold testing, and evoked potential testing to evaluate peripheral nervous system involvement in 40 patients with GA. All the patients tested were homozygotes or compound heterozygotes for one single OAT mutation (L402P). Fifty-three percent of the patients had at least two abnormal findings on neurography, prolonged F-latency being the earliest detectable change characteristic of sensorimotor axonal distal neuropathy. None of the patients studied had disabling neuropathy, 40% of the patients had mild and asymptomatic neuropathy, and 10% had symptomatic peripheral neuropathy. The neurography findings correlated with the severity of GA fundus changes, and patients with signs of neuropathy had the most severe ophthalmologic GA stage; Cr supplements had no effect on neuropathy prevalence.
Skeletal muscle: McCulloch and Marliss34 were the first to report atrophy of, and the presence of tubular aggregates in, type 2 skeletal muscle fibers of one patient with GA (Fig. 3), a finding later confirmed by various other investigators.35,38,39,40 All reported abnormalities have been confined to type 2 fibers, type 1 fibers being normal in appearance and number in all examined biopsy specimens reported to date. Similar histologic muscle changes were noted in the iris dilator muscle of one patient.41 Tubular aggregates appear to be more common in GA than in any other disease, and aside from patients with GA, they are rarely observed in female patients. They have been noted in male patients in periodic paralysis, hyperthyroidism, porphyria cutanea tarda, acromegaly, myasthenia gravis, polymyositis, myotonic dystrophy, muscular viral infections, denervations, and drug and alcohol abuse, and in some apparently normal persons. The reason that female patients with these disorders do not have tubular aggregates is unknown. Tubular aggregates are thought to represent a detoxifying mechanism or a response to injury and may perhaps be a result of muscle degeneration, eventually disappearing with the affected fiber. Consistent with this hypothesis is the finding by Sipila and colleagues39 of nonspecifically abnormal electromyograms in 16 of 17 deltoid muscle recordings in GA patients, indicative of a mild to moderate skeletal muscle myopathy in GA. The progression of the myopathy is slower than that of the chorioretinopathy, but muscle is nevertheless fairly severely affected by the underlying metabolic defect. Approximately 10% of GA patients demonstrate mild proximal muscle weakness.8 Kaiser-Kupfer and co-workers,35 who found tubular aggregates in skeletal muscle biopsies of three of four patients, grew the muscle cells of these patients in tissue culture. The addition of 20 mmol/L ornithine to the culture media was lethal to GA muscle cells within 48 to 72 hours, but had no effect on cultured normal muscle cells. In addition to type 2 muscle fiber atrophy and tubular aggregates, Valtonen and colleagues43 found computed tomography and MRI changes in the thigh muscles of seven GA patients, although relaxation time measurements gave little additional muscle metabolism insights. Heinanen and colleagues,44 in conjunction with studying brain Cr,36,37 studied the creatine phosphate (CrP) levels in skeletal muscle of GA patients. Abnormal 31 P-MR spectra were found in resting calf muscle of GA patients, with decreased ratios of PCr/adenosine triphosphate (ATP) and PCr/Pi, proposed to reflect a decreased CrP concentration in calf muscle of GA patients. However, these decreases did not reflect the clinical stage of the disease, degree of muscle destruction, or ornithine concentrations. The researchers proposed that disturbed cellular energy production resulting from intracellular ornithine-induced inhibition of Cr synthesis, and decreased PCr muscle stores may partially explain the pathogenesis of GA. Daily oral supplementation with physiological levels of Cr (1.5–2 g/day) for 8 to 15 years led to almost normalized PCr/P and PCr/ATP ratios in the calf muscle of GA patients, likely by increasing intracellular PCr.45

Fig. 3. A. A muscle fiber of a patient with gyrate atrophy shows a large inclusion. B. On high power, the inclusion is composed of tubular aggregates.
Liver: Liver biopsies in patients with GA have demonstrated bizarre, elongated, and segmented mitochondria—an abnormality perhaps similar to that seen in the hyperornithinemia, hyperammonemia, and homocitrullinuria (HHH) syndrome (see below; Fig. 4). These changes are thought to be a direct result of hyperornithinemia, since similar changes have been produced in rats maintained on diets high enough in ornithine content to produce a constant moderate hyperornithinemia.46 Fibroblasts from GA patients, as well as normal fibroblasts exposed to high levels of ornithine in the growth media, exhibit similar mitochondrial changes in tissue culture.47 Similar mitochondrial changes have also been observed in skeletal muscle of GA patients.41
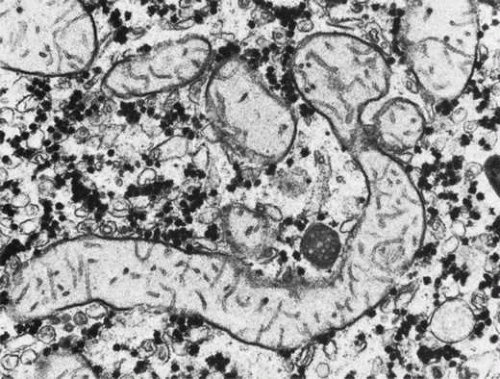
Fig. 4. An elongated mitochondrion in a liver cell from a patient with gyrate atrophy. (Courtesy of M.J. Phillips, MD.)
Hair: Ten patients with GA have been noted to have peculiar, fine, straight, occasionally sparse scalp hair with areas of alopecia. Scanning electron microscopy revealed that the medullary portions of sampled hairs contained loosely packed macrofibrils. The intervening spaces appeared to be packed with a structureless, electronlucent compact substance that was insoluble in both water and common laboratory solvents. The exact nature of this substance and the relationship of this finding to the oculometabolic disease remain obscure.35
Other: Francois48 reported an association of Alder’s anomaly (the presence of numerous azurophilic granules in the cytoplasm of neutrophils, which stain dark violet by Pappenheim’s technique) with GA in one patient. However, Alder’s anomaly has generally been associated with Hurler syndrome and has not been reported in association with GA by other investigators. In a study performed at the U.S. National Eye Institute, and therefore not expected to deal with a homogenous genetic population (as one might find with Finnish GA patients), an increased occurrence of thyroid disease in patients with GA compared with control patients was found by Whitcup and colleagues.49 Seven of 34 patients with GA had thyroid disease, resulting in an estimated odds ratio for thyroid disease in GA patients of 12.7 that of normal controls. Similar but less pronounced results were found in retinitis pigmentosa patients, with an estimated odds ratio of 6.2 compared with normal controls.
ANIMAL DATA
Hepatic mitochondrial changes similar to those seen in patients with GA have been produced in normal rats maintained on high-ornithine diets.50 A case of an adult male cat with retinal degeneration, extreme hyperornithinemia, and absence of OAT—an apparent replica of human GA—was reported by Valle and associates.51 Attempts to breed the cat were unsuccessful, and it later died. Histopathologic examination of the cat’s eyes demonstrated extensive neuroretinal and RPE damage and cell loss. There was a decrease in the number of small choroidal vessels, but not the large ones. In cats with other retinal degenerations, such loss of RPE and choriocapillaris is uncommon. This “GA cat” did not have cataracts.
Kuwabara and associates52 demonstrated selective destruction of RPE cells in rats and monkeys after intravitreal injections of ornithine. Secondary destruction of overlying photoreceptors and underlying choroid developed later. Based on these data, they suggested that the RPE may be the primary target organ in GA. A more recent attempt by Daune-Anglard and colleagues53 to mimic the damage of GA with induced ornithine toxicity in mice and chickens using 5-fluoromethylornithine (5FMOrn) administered orally for 53 days showed no ocular pathologic changes in these animals despite a tenfold increase in tissue ornithine. However, 10% to 20% of tissue OAT is refractory to inactivation by 5FMOrn, so these results only show that mice and chickens will not develop GA as a result of sustained hyperornithinemia due to incomplete blockage of OAT for this period of time.
Subsequently, Wang and colleagues54 successfully created a mouse model of GA that closely mimics GA in human patients. Using targeted disruption of the murine OAT gene, they produced OAT-deficient mice that exhibit chronic hyperornithinemia, ten to 15 times normal. These OAT-deficient mice developed slowly progressive retinal degeneration over the first 12 months of life. The RPE cells were the initial site of damage. Using this mouse model of GA, Wang and colleagues55 further evaluated the effect of long-term reduction in ornithine on prevention of retinal degeneration. OAT-deficient mice fed an arginine-restricted diet for 12 months had significantly reduced plasma ornithine levels. Importantly, retinal degeneration, as measured by ERG and retinal histologic and ultrastructural studies, was prevented. The researchers concluded that ornithine accumulation, and not OAT activity in the retina and RPE, is a necessary factor in the pathophysiology of retinal degeneration in GA.
DIFFERENTIAL DIAGNOSIS
Ocular
Although cases of GA have been mislabeled as many other things, usually “atypical retinitis pigmentosa,” and patients referred as having GA often turn out to be suffering from some other disorder, the diagnosis is actually quite easy to make if the five main features (see previous Diagnosis section) can be recalled (Fig. 5). Confusion in diagnosis is primarily due to the rarity of the disease. It is extremely unlikely that any primary care ophthalmologist would have the opportunity to make this diagnosis more than once in a career, since the incidence of GA appears to be less than one in 1,000,000 everywhere except in Finland, where it occurs in about one in 50,000 individuals.8
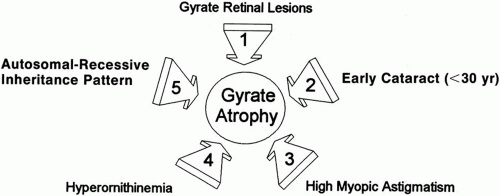 Fig. 5. The five main features in the diagnosis of gyrate atrophy. |
Almost invariably, the main ophthalmic features of high myopic astigmatism, posterior subcapsular cataracts, and typical retinal appearance are obvious to the ophthalmologist. One can be quite certain that if the patient does not have the typical fundus picture, as well as extreme hyperornithinemia, GA is not the correct diagnosis. The autosomal recessive inheritance pattern can usually be quickly ascertained with a brief family history asking about affected relatives and parental consanguinity (see Fig. 5), even before confirmatory laboratory biochemical and genetic testing is undertaken.
Non-GA patients with extremely high myopia (usually more than -15 D) may occasionally have posterior polar, peripapillary, or peripheral clusters of round, full-thickness, chorioretinal atrophic lesions, often causing significant reduction in visual acuity.56 This, however, is not the pattern of the lesions seen in GA.
An entity of “central gyrate atrophy” has been described,57 but this is probably equivalent to end-stage serpiginous choroidopathy rather than a separate inherited chorioretinopathy.58,59
Large areas of paving stone degeneration may superficially resemble GA, albeit on a much smaller scale. Paving stone degeneration is usually found in the inferior quadrants peripherally, whereas GA is not segmental and involves all 360 degrees of the fundus.60 Occasional reports of atypical GA appear in the literature.61,62,63,64 Review of these cases usually reveals either that the retina did not quite have the typical appearance of GA, myopia and cataracts were not present, and/or hyperornithinemia was absent. It is interesting to speculate as to why other disorders have retinal lesions similar in appearance to GA and whether or not some final common pathway to chorioretinal destruction exists, especially in cases where some similar systemic involvement may coexist, as in muscular dystrophy. However, labeling such cases with similar lesions as “atypical gyrate atrophy” can be misleading, especially to the majority of practitioners who have never encountered GA, and may lead to corruption of research efforts and reports by the inclusion of different etiologic disorders in supposedly homogeneous study groups.65
Metabolic
Hyperornithinemia is not unique to GA.11,46 It was reported in two other conditions. In the late 1960s, Kekomaki and colleagues66 and Bickel and associates67 described two siblings who presented with failure to thrive, prolonged neonatal jaundice, atypical hepatic cirrhosis, renal tubular dysfunction, and mental retardation. Hyperornithinemia (about three times normal), renal glycosuria, generalized aminoaciduria, and mild hyperammonemia were present. Hepatic OAT levels were about one-sixth those of normal patients. Garnica and coworkers68 later confirmed these findings. Kekomaki and coworkers66 studied the enzyme kinetics of the residual OAT in the hepatocytes of one patient and found them to be normal, suggesting that the problem in this disorder is one of decreased enzyme synthesis or excessive degradation. Both siblings, at ages 15 and 9, were severely retarded, had normal ocular examinations, normal plasma ornithine, normal liver function tests, generalized aminoaciduria, elevated serum creatinine, and hypertension. The etiology of this syndrome remains unknown.8
The second disorder with associated hyperornithinemia is HHH syndrome, a rare autosomal recessive disorder that is about one-third as common as GA. HHH patients exhibit hyperornithinemia about one-half as elevated as that of GA patients, but there is considerable overlap. In the late 1960s to early 1970s, Shih and colleagues69,70 and later (1975) Gatfield and colleagues71 described patients presenting in infancy with feeding difficulty, mental retardation, and seizures, in which the biochemical triad of hyperornithinemia, hyperammonemia, and homocitrullinuria was observed. Abnormal hepatic mitochondria, not unlike those seen in GA, have been identified in this disease. The biochemical defect was hypothesized by Fell and coworkers72 to reside in defective transport of ornithine across the inner mitochondrial membrane into the mitochondrial matrix, and the biochemical and clinical evidence supports this hypothesis.8,73,74,75,76,77,78,79,80,81,82 The ornithine/citrulline transporter was identified and confirmed as the defect in the HHH syndrome.83,84,85 The gene has been labeled ORNT1 and mapped to 13q14.86 HHH patients may voluntarily restrict their protein intake to avoid having symptoms. The symptoms are thought to be due to hyperammonemia as they are similar to other hyperammonemia syndromes, and can be dramatically ameliorated with ornithine HC1, 0.5 to 1.0 mmol/kg/day dietary supplementation.78 The additional dietary ornithine is thought to elevate cytosolic ornithine levels, thereby driving it into the mitochondrial matrix, where it acts as the limiting substrate in the elimination of ammonia via the urea cycle. HHH syndrome, like GA, is inherited as an autosomal recessive disorder. Some patients do not respond to ornithine or arginine supplementation, suggesting heterogeneity in the mutation of the affected protein. Ocular examination has been normal in all patients examined.8
In neither of the above two metabolic syndromes have ocular abnormalities been noted. The senior author (SAA) repeatedly examined a currently 26-year-old patient and a teenage patient, both with HHH syndrome, as they underwent ornithine supplementation treatment over many years. Neither patient manifested any retinal or ERG changes (Steve A. Arshinoff, unpublished data, York Finch Eye Associates, Toronto, Ontario, Canada, 1985–2003). GA retinal lesions have not been observed in an infant. Hayasaka and coworkers19 noted that a 2-year-old patient with biochemical GA did not develop fundus lesions until age 4, and Stoppoloni and colleagues87 reported on a 13-year-old patient who had early fundus lesions. The senior author has followed a GA patient, who first presented at age 7 with well-defined peripheral GA lesions involving 360 degrees of her retina.
THE METABOLISM OF GYRATE ATROPHY
ALTERED PLASMA AMINO ACID PROFILE
Ornithine is a dibasic amino acid that is not normally found in proteins (Figs. 6 and 7). However, it plays a pivotal role in the urea cycle (Fig. 8). The initial discovery by Simell and Takki9 of extreme elevations of ornithine levels in the plasma, urine, cerebrospinal fluid, and aqueous humor of patients with GA encouraged others to search for secondary alterations in amino acids. McCulloch and coworkers11 and Arshinoff and coworkers46 reported the presence of hypolysinemia, hypoglutamic acidemia, and hypoglutaminemia, which was confirmed by Valle and coworkers,88 who also found the presence of hypoammonemia. The mechanism of the hypolysinemia, hypoglutamic acidemia, hypoglutaminemia, and hypoammonemia has been proposed to be excessive excretion secondary to renal tubular readsorption competitive blockade by hyperornithinuria in the renal proximal tubular filtrate.46,88,89
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


