 Glial Tumors of the Retina and Optic Disc
Glial Tumors of the Retina and Optic DiscPseudoneoplastic Gliosis of the Retina
General Considerations
Lesions that originate from retina glial cells include reactive gliosis, nodular pseudoneoplastic gliosis, astrocytic hamartoma, and acquired retinal astrocytoma. Reactive gliosis is common and usually appears as a sheetlike lesion that does not simulate a neoplasm. Macular surface wrinkling retinopathy is a classic example. Nodular pseudoneoplastic gliosis can sometimes simulate an intraocular tumor (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11).
Pseudoneoplastic gliosis (“massive gliosis”) of the retina is a proliferation of glial cells that attains tumorous proportions. We prefer not to use the term massive gliosis for all such cases because many are small retinal nodules that are not massive. Pseudoneoplastic gliosis occurs most often in eyes with chronic inflammation, prior ocular trauma, Coats disease, retinal hemangioblastoma, or congenital malformations. In some cases, it is not diagnosed clinically but is recognized histopathologically after enucleation of a blind, uncomfortable eye (1, 2, 3). It might have clinical and histopathologic features similar to those of the secondary vasoproliferative tumor described in the prior chapter. Based on the available information, however, we continue to classify the entities separately.
Clinical Features
Pseudoneoplastic gliosis can occur in an eye with clear media and no prior ocular insults. In such instances, it appears ophthalmoscopically as a solitary, white-yellow, superficial retinal lesion. Yellow intraretinal and subretinal exudation can occur. In other instances, it develops in an area of chronic retinal detachment or on a scleral buckle from retinal detachment surgery. Small foci of retinal gliosis have been observed spontaneously to resolve (9,10). Pseudoneoplastic gliosis has been recognized to appear spontaneously and show rather rapid growth (10). In eyes with opaque media, the lesion may be detected coincidentally with ultrasonography. In those instances, it might be ultrasonographically similar to other intraocular neoplasms.
Diagnostic Approaches
There are no specific findings with ancillary diagnostic studies such as fluorescein angiography or ultrasonography. However, the diagnosis can be suspected based on ocular history and ophthalmoscopic findings. With fluorescein angiography, it has similar features to vasoproliferative tumor and other glial tumors described later in this chapter. With ultrasonography, the mass usually shows medium to high internal reflectivity. It is usually sessile or dome shaped and is not known to assume a mushroom configuration.
Pathology
Histopathologically, pseudoneoplastic gliosis consists of a mass of closely arranged, well-differentiated astrocytes (2,11). It is usually markedly vascular, raising the possibility that some cases might represent a primary vascular tumor with secondary gliosis. Dystrophic calcification and even bone formation can be present (2). A nodule of gliosis has been found histopathologically in a blind eye with retinitis pigmentosa (5). Microscopically, pseudoneoplastic gliosis can be similar to a pilocytic astrocytic hamartoma or acquired retinal astrocytoma.
Management
Because pseudoneoplastic gliosis is not usually diagnosed clinically, there are no established guidelines regarding management. If the diagnosis is strongly suspected based on clinical findings in an asymptomatic patient, then periodic observation is warranted. If such a lesion shows progression with retinal detachment and exudation, then it can be managed according to the clinical findings. This can involve laser photocoagulation, cryotherapy, radiotherapy, or local resection. If massive gliosis is suspected in a blind phthisical, otherwise asymptomatic eye, no specific treatment is necessary. In such a case, however, enucleation might be justified if malignancy is a legitimate possibility.
Selected References
1. Shields JA, Shields CL. Glial tumors of the retina and optic disc. In: Shields JA, Shields CL, eds. Intraocular Tumors. A Text and Atlas. Philadelphia: WB Saunders; 1992:421-435.
2. Yanoff M, Zimmerman LE, Davis RL. Massive gliosis of the retina. Int Ophthalmol Clin 1971;11:211-229.
3. Nowinski T, Shields JA, Augsburger JJ, et al. Exophthalmos secondary to massive intraocular gliosis in a patient with a colobomatous cyst. Am J Ophthalmol 1984;97:641-643.
4. Gelisken F, Inhoffen W, Rohrbach JM, et al. Massive retinal gliosis: a late complication of retinal detachment surgery. Graefes Arch Clin Exp Ophthalmol 2004;242:255-258.
5. Rodrigues MM, Bardenstein D, Wiggert B, et al. Retinitis pigmentosa with segmental massive retinal gliosis. An immunohistochemical, biochemical, and ultrastructural study. Ophthalmology 1987;94:180-186.
6. Berger B, Peyman GA, Juarez C, et al. Massive retinal gliosis simulating choroidal melanoma. Can J Ophthalmol 1979;14:285-290.
7. Green WR. Bilateral Coats’ disease. Massive gliosis of the retina. Arch Ophthalmol 1967;77:378-383.
8. Ryan H. Massive retinal gliosis. Trans Ophthalmol Soc Aust 1954;14:77-83.
9. Demirci H, Shields JA, Shields CL, et al. Spontaneous disappearance of presumed retinal astrocytic hyperplasia. Retina 2002;22:237-239.
10. Khawly JA, Matthews JD, Machemer R. Appearance and rapid growth of retinal tumor (reactive astrocytic hyperplasia?). Graefes Arch Clin Exp Ophthalmol 1999;237:78-81.
11. Ganley JP, Streeten BW. Glial nodules of the inner retina. Am J Ophthalmol 1971;71:1099-1103.
▪ Pseudoneoplastic Gliosis of the Retina
Pseudoneoplastic (reactive) gliosis of the retina can assume a variety of clinical and histopathologic appearances. Focal gliosis is generally a dense yellow-white or gray lesion that is different from most astrocytic hamartomas of tuberous sclerosis complex.
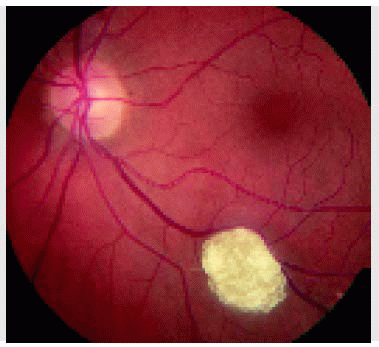
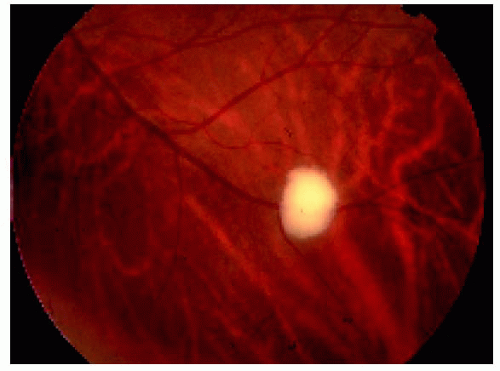 Figure 21.1. Pseudoneoplastic gliosis of the retina. This yellow-white superficial retinal nodule in a healthy 51-year-old man has been stable for several years. The etiology is unknown. |
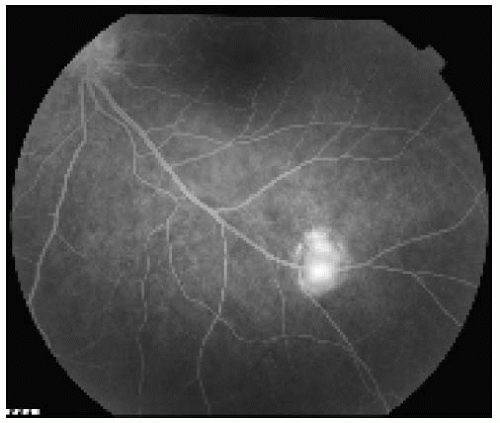 Figure 21.2. Fluorescein angiogram in the late venous phase of the lesion shown in Figure 21.1, revealing fairly intense hyperfluorescence of the lesion. The fluorescence of the lesion began in the early venous phase. |
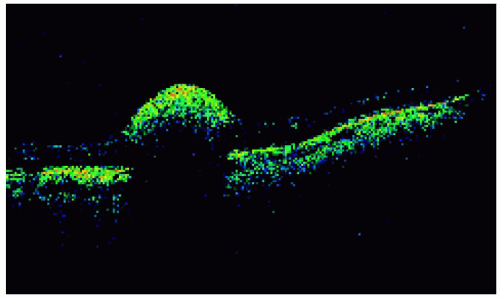 Figure 21.3. Optical coherence tomography of the lesion shown in Figure 21.1. Note the shadowing of the deep retina and choroid due to the highly reflective, superficial lesion. |
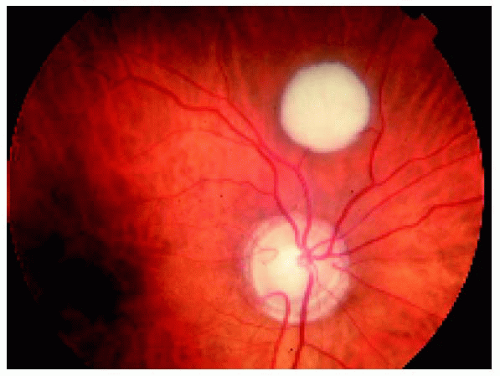 Figure 21.4. Presumed reactive gliosis of the retina located superior to the optic disc in the right eye of a 45-year-old woman. The optic disc has a congenital abnormality, perhaps a mild variant of coloboma. |
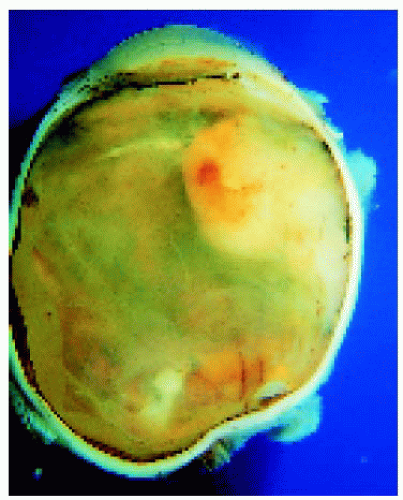 Figure 21.5. Mass of reactive gliosis located in peripheral fundus of an eye enucleated for chronic discomfort many years after surgery for congenital cataract followed by numerous complications. |
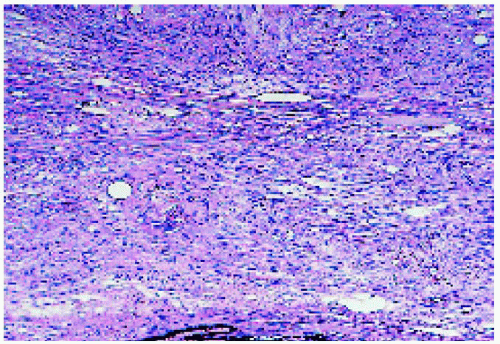 Figure 21.6. Histopathology, showing a mass composed of well-differentiated glial cells. (Hematoxylin-eosin ×100.) |
Retinal Astrocytic Hamartoma
General Considerations
Retinal astrocytic hamartoma is a benign retinal tumor that is composed of glial cells, predominantly astrocytes (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26). It is believed to be congenital in most cases, but it can become clinically apparent sometime after birth. It does not metastasize. It is frequently associated with tuberous sclerosis complex (TSC), a syndrome that includes various combinations of low-grade intracranial astrocytoma, cutaneous angiofibromas (“adenoma sebaceum”), cutaneous depigmented macules, cardiac rhabdomyoma, renal angiomyolipoma, and other hamartomas (2,3). In those cases that are part of TSC, various genetic alterations have been identified on chromosomes 9 and 16. Some patients have only the retinal tumor without additional findings of TSC. It is undetermined whether they represent a separate entity or a forme fruste, or partial expression, of TSC. An identical fundus tumor is occasionally seen in patients with neurofibromatosis type 1.
Clinical Features
Ophthalmoscopically, retinal astrocytic hamartoma can show considerable variation from case to case. The two most common variations are the noncalcified tumor and the calcified tumor. The noncalcified variant appears as a gray-yellow, sessile lesion in the inner aspect of the sensory retina. It can be transparent and fairly flat, sometimes suggesting retinal gliosis. Slightly larger lesions have a gray-yellow color and may cause retinal traction. The calcified variant may have minimal calcification or may be totally calcified. The characteristic feature is glistening yellow spherules of calcification. This glistening calcification differs from the duller, chalky calcification that characterizes retinoblastoma. Many lesions show both calcified and noncalcified components.
In contrast to retinoblastoma, astrocytic hamartoma does not usually develop prominent retinal feeding and draining blood vessels. It often is associated with mild to moderate retinal traction, a finding not usually seen with a comparable-sized untreated retinoblastoma. Occasionally, the tumor appears as a deep retinal lesion that is usually noncalcified and can resemble subretinal fibrosis.
Although astrocytic hamartoma is usually a relatively stable lesion, it can show progressive growth and exhibit locally malignant behavior. We have seen cases that showed progressive growth, exudative retinal detachment, and neovascular glaucoma, ultimately requiring enucleation. Extraocular extension into the orbital and epibulbar tissues has been recognized in these cases (20, 21, 22).
Diagnostic Approaches
Retinal astrocytic hamartoma, particularly the calcified variant, usually shows autofluorescence (9). Fluorescein angiography of the typical lesion shows a characteristic network of small blood vessels in the venous phase with fairly intense late staining. In the case of a calcified lesion, ultrasonography shows a calcified plaque as might be seen with choroidal osteoma or calcified retinoblastoma. Optical coherence tomography (OCT) can be used to document the superficial location of the lesion and its highly reflective features. Cytopathologic study of fine needle aspiration biopsy can be employed to make the diagnosis in atypical cases (7).
Pathology
Histopathologically, astrocytic hamartoma is usually composed of elongated fibrous astrocytes that have small uniform nuclei and interlacing cytoplasmic processes. Areas of calcification may be present, often in the form of calcospheres (5). Some larger tumors may contain moderately pleomorphic gemistocytic astrocytes. The less common, locally invasive variant is generally located on the optic disc and has large, poorly differentiated cells similar to the subependymal astrocytomas that are seen in the brain in some patients with TSC (20,21).
Management
The majority of astrocytic hamartomas are small, extrafoveal, stationary lesions, with little or no tendency to grow or cause complications. However, they should be followed periodically because some can show progressive growth, exudative retinal detachment, and neovascular glaucoma. When a lesion is growing and is suspected to have potential for such proliferation, then treatment is warranted. Depending on the circumstances, laser photocoagulation, cryotherapy, or vitrectomy and retinal detachment surgery may be necessary. Most cases of the giant cell variant have come to enucleation because of neovascular glaucoma. However, we believe that if such a tumor is detected early, treatment with irradiation or other methods might possibly achieve tumor control and avoid enucleation.
Selected References
1. Shields JA, Shields CL. Glial tumors of the retina and optic disc. In: Shields JA, Shields CL, eds. Intraocular Tumors. A Text and Atlas. Philadelphia: WB Saunders; 1992:421-435.
2. Shields JA, Shields CL. Systemic hamartomatoses (“phakomatoses”). In: Shields JA, Shields CL, eds. Intraocular Tumors. A Text and Atlas. Philadelphia: WB Saunders; 1992:513-539.
3. Nyboer JH, Robertson DM, Gomez MR. Retinal lesions in tuberous sclerosis. Arch Ophthalmol 1976;94:1277-1280.
4. Shields JA, Shields CL. The systemic hamartomatoses (“phakomatoses”). In: Nelson LA, Olitsky SE, eds. Harley’s Pediatric Ophthalmology, 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2005:436-447.
5. Margo CE, Barletta JP, Staman JA. Giant cell astrocytoma of the retina in tuberous sclerosis. Retina 1993;13:155-159.
6. Mullaney PB, Jacquemin C, Abboud E, et al. Tuberous sclerosis in infancy. J Pediatr Ophthalmol Strabismus 1997;34:372-375.
7. Shields JA, Shields CL, Ehya H, et al. A typical retinal astrocytic hamartoma diagnosed by fine-needle biopsy. Ophthalmology 1996;103:949-952.
8. Zimmer-Galler IE, Robertson DM. Long-term observation of retinal lesions in tuberous sclerosis. Am J Ophthalmol 1995;119:318-324.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


