Glaucoma Associated with Corneal Disorders
Paula Anne Newman-Casey
Joshua D. Stein
Glaucoma is associated with various corneal abnormalities; it also can develop as a result of medical or surgical interventions used to treat corneal disease. This chapter describes some of the challenges associated with detecting and monitoring glaucoma in patients with corneal disorders; describes several corneal disorders known to be associated with glaucoma; and discusses glaucoma that develops after corneal surgical procedures, such as penetrating keratoplasty (PKP) and lamellar keratoplasty. Developmental abnormalities of the anterior segment may affect both the cornea and chamber angle, as in patients with the Axenfeld–Reiger syndrome or Peters anomaly; these conditions are discussed elsewhere in this series. Developmental abnormalities of the anterior segment may affect both the cornea and chamber angle, as in patients with the Axenfeld–Reiger syndrome or Peters anomaly are beyond the scope of this chapter. These conditions are discussed elsewhere in this series.
Identifying Glaucoma in Patients with Corneal Disease
Monitoring glaucoma status can be challenging in patients who have comorbid ocular conditions that affect the integrity or clarity of the cornea. Corneal edema, corneal scarring, corneal surface irregularities, and significant corneal astigmatism can all affect the accuracy of intraocular pressure (IOP) measurements obtained by using Goldmann applanation tonometry. In addition, because it is now well appreciated that central corneal thickness can affect the accuracy of IOP measurements, clinicians should consider performing pachymetry when evaluating a patient with a corneal condition for glaucoma.1 Clinicians may significantly underestimate IOP in patients who have corneal conditions associated with corneal thinning, and the falsely low IOP measurements in these patients may result in a delay in the recognition of glaucoma. In contrast, in patients with increased central corneal thickness, the IOP may be overestimated; clinicians who do not consider the potential effect of corneal thickness on the IOP reading might initiate pressure-lowering interventions unnecessarily. In the immediate period following PKP, the presence of corneal edema, surface irregularities, and astigmatism make obtaining an accurate IOP measurement by using Goldmann applanation tonometry nearly impossible.2 In patients who have corneal disease or who have undergone PKP, other methods, including pneumotonometry, the Tono-Pen, and the Mackay-Marg tonometer, may yield more-accurate readings.
Evaluating for glaucoma can be difficult, as well, because the underlying corneal disorder can affect the clinician’s ability to perform structural and functional testing to assess for glaucomatous damage. Corneal disorders can limit the gonioscopic view of angle structures to detect the mechanism of the glaucoma—such as the presence of peripheral anterior synechiae (PAS) in patients with infectious or inflammatory corneal disorders, or the presence of angle recession in patients with corneal scarring from trauma. Although topical glycerin can be used to reduce corneal edema and facilitate visualization of angle structures in some patients, unfortunately, in many patients with corneal pathology, the clinician cannot adequately evaluate the angle.
Often when corneal pathology compromises visual acuity, performing visual field testing becomes difficult. Traditional testing for glaucoma by using standard automated perimetry often requires visual acuity of better than 20/400. In patients with vision of 20/400 or worse, Goldmann field testing can be attempted. Nguyen et al3 showed that frequency-doubling perimetry is not significantly adversely affected by corneal topographic changes; this may be an alternative means of helping detect glaucoma in some patients who have corneal abnormalities or have undergone keratoplasty. Finally, corneal disorders also may limit evaluation of the optic nerve and retinal nerve fiber layer.
In the past, IOP was considered the key factor in determining whether a patient had glaucoma. However, recent studies have demonstrated that many patients with ocular hypertension never develop glaucoma, whereas at least 30% of patients with glaucoma never have IOPs greater than 21 mm Hg.4 Therefore, although elevated IOP is an important risk factor for glaucoma, it is not part of the disease definition. Thus, when evaluating a patient with corneal pathology for glaucoma, it is important, whenever possible, to focus on the status of the optic nerve and retinal nerve fiber layer, along with results of perimetry, and to give less attention to IOP readings, which may be less accurate.
Glaucoma Associated with Primary Corneal Endothelial Disorders
Iridocorneal Endothelial Syndrome
The iridocorneal endothelial (ICE) syndrome is a unilateral, nonheritable disease characterized by abnormalities of the corneal endothelium, iris, and anterior segment. ICE syndrome is most common in white women in the third to fifth decades of life. The abnormal corneal endothelial cells, called ICE cells, form a membrane that migrates from the posterior surface of the cornea across the trabecular meshwork and onto the surface of the iris.5,6 The resulting clinical manifestations include corneal edema, iris changes, PAS, and glaucoma. Historically, the disease was categorized into three subtypes, mainly on the basis of the appearance of the iris: Chandler syndrome, progressive (essential) iris atrophy, and Cogan-Reese (iris-nevus) syndrome.7 More recently, the three variants of ICE syndrome have come to be seen as a spectrum of clinical presentations of the same disease.
Clinical Features
Patients with ICE syndrome can present with unilateral decreased visual acuity, halos, or with ocular discomfort due to corneal edema or elevated IOP.8,9 The diagnosis of ICE syndrome is made if the clinical findings satisfy two of the following three criteria: (i) typical unilateral hammered-silver appearance of the posterior cornea or corneal edema so severe that evaluation of the posterior cornea is impossible; (ii) iris atrophy with corectopia, iris holes, iris nodules, or ectropion uvea in the eye with the corneal changes; (iii) PAS. The iris atrophy can lead to large and sometimes bizarrely shaped iris “stretch” holes, which are caused by traction between the iris and PAS on opposite sides of the globe (Fig. 54.1).5,8,10 Less commonly, smaller oval holes may develop adjacent to PAS. These “melting” holes are presumed to be caused by iris ischemia resulting from obstruction of iris vessels by the synechiae.5,8 Pigmented lesions that range from multiple, pedunculated, nodular lesions to a more diffuse, smooth, velvety change of the iris can form when the membrane composed of ICE cells encircles and pinches off” portions of the iris.5,6,11 In addition, Odenthal et al12 have suggested that the ICE tissue has dysfunctional corneal pump activity, resulting in the development of corneal edema.
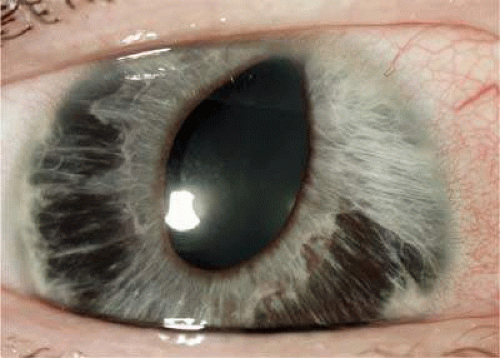 Figure 54.1. Slitlamp photograph of a patient with ICE syndrome. The iris demonstrates diffuse areas of stromal atrophy, and the pupil is irregular and dragged superiorly. |
Histopathology
Using specular, electron, and confocal microscopy, investigators have carefully studied the abnormal ICE cells responsible for the beaten metal appearance of the posterior cornea.5,6 Specular microscopy reveals that the ICE cells are relatively large and pleomorphic; the examination also shows that the cells have a specular reflex with light-dark reversal, in which the surface is dark and the intercellular borders are light and indistinct.13 (Fig. 54.2). Scanning electron microscopy and transmission electron microscopy demonstrate that ICE cells resemble epithelial cells more closely than they do endothelial cells.14 The ICE cells become confluent and show four basic patterns: total ICE, in which no normal endothelial cells are seen; disseminated ICE, in which ICE cells are scattered throughout the mosaic of enlarged endothelial cells; subtotal ICE plus, in which an area of abnormal ICE tissue is sharply demarcated from the abnormally small endothelium; and subtotal ICE minus, in which an area of ICE tissue merges with abnormally large endothelial cells.15 In vivo confocal microscopy can be used to visualize these groups of epithelial-like cells on the endothelium.13,16 Using confocal microscopy, the ICE cells appear to have epithelial-type characteristics including hyperreflective nuclei with cobblestone-like central elevation and saclike blisters, and the cells become increasingly epithelioid and pleomorphic as the disease progresses. This is especially helpful in patients whose advanced corneal edema precludes the use of traditional specular microscopy for diagnostic purposes. In patients with significant corneal edema that limits visualization of the iris and angle structures, ultrasonographic biomicroscopy can be useful for detecting PAS and iris atrophy, to aid in establishing the diagnosis of the ICE syndrome.17
 Figure 54.2. Specular microscopy of the corneal endothelium in ICE syndrome. Cell borders are obscured, resulting in loss of the normal endothelial mosaic. Note dark areas within endothelial cells. Brighter reflections are believed to be from cell borders. |
Pathophysiology
Historically, researchers had hypothesized that the ICE cells are epithelial cells that may have been misplaced during embryonic development. Now, however, it is thought instead that endothelial cells undergo a metaplastic transformation during which they acquire a partial phenotype of an epithelial cell. Microscopy reveals that these cells have proteins characteristic of epithelial and endothelial cells; and immunohistochemistry reveals that the ICE cells stain for both epithelial keratin and endothelial intermediate filaments.18,19,20 Furthermore, these cells exhibit pleomorphism and loss of contact inhibition, similar to cells undergoing a neoplastic process.21
Some researchers have suggested that viruses may play a role in the pathogenesis of ICE syndrome. Herpes simplex virus (HSV) DNA has been identified on the corneal endothelium and in the aqueous humor in some patients with ICE syndrome; another study reported that Epstein-Barr virus (EBV) titers were elevated in a series of patients with the syndrome.22,23,24 It remains unclear whether a virus might trigger the development of ICE syndrome or whether viruses such as HSV and EBV are more easily reactivated in patients with ICE syndrome.25
Iridocorneal Endothelial Syndrome and Glaucoma
The membrane of ICE cells can migrate across the trabecular meshwork and obstruct the anterior chamber angle, causing secondary angle-closure glaucoma. Studies have shown that 42% to 82% of patients with the ICE syndrome develop secondary glaucoma, and those patients with progressive iris atrophy tend to develop a more severe form of glaucoma.15,26,27
In the early stages of the disease process, before extensive PAS occlude the filtration angle, hypotensive medications can be used to control glaucoma in patients with the ICE syndrome. Because angle closure is often the mechanism responsible for the elevated IOP in these patients, medications that decrease aqueous production are more useful than miotics, which increase outflow through the trabecular meshwork. Laser trabeculoplasty generally is ineffective for the same reason. As ICE syndrome is a progressive disease, medication use often becomes ineffective for IOP control over time; thus, in patients who have ICE syndrome and glaucoma, surgical intervention may eventually be required.15,28
The two most common surgical interventions for glaucoma are trabeculectomy and glaucoma drainage-device (GDD) implantation. Although both these procedures can reduce IOP, their success rates are lower among patients with the ICE syndrome and glaucoma than among persons with primary open-angle glaucoma or other forms of glaucoma; this is because ICE cells can continue to proliferate and block the trabeculectomy sclerostomy or grow over the tip of the GDD.28 In a study by Lanzl et al,28 8 of 10 patients with the ICE syndrome who had received mitomycin C during trabeculectomy experienced surgical success (mean follow-up, 15 months), whereas only 13 of 22 patients in a series of similar patients had success after undergoing trabeculectomy without the use of an antimetabolite (mean follow-up, 12 months). These authors have postulated that mitomycin C may decrease aqueous production and inhibit ICE cell proliferation. The efficacy of adjunctive use of 5-fluorouracil is more controversial.26,27,29 Reported success rates with the use of drainage devices in patients with the ICE syndrome have ranged from 71% to 90% at 1 year and 53% to 75% at 5 years.26,27 Among a group of 21 patients with the ICE syndrome who had glaucoma and underwent implantation of a GDD, there was a significant increase in glaucoma drainage implant survival with the use of a Baerveldt implant (11/13 successes) when compared with the Molteno device (2/7 successes) (mean follow-up 50 months).27 Patients with ICE syndrome who receive GDDs may require one or more surgical revisions if ICE cells occlude the tip of the tube; to facilitate future tube repositioning, it has been suggested that the tube should be cut longer than usual. When trabeculectomy and GDD implants cannot control the IOP, performing cyclodestructive procedures may be necessary.
Management of Corneal Edema
When corneal edema becomes visually significant, hypertonic saline eye drops and ointment can be used initially to attempt to reduce corneal swelling. Some clinicians believe the use of aqueous suppressants to decrease IOP may lead to an improvement in corneal edema, although the effectiveness of this approach has been inadequately studied. Once corneal edema becomes refractory to medical management and leads to reduced visual acuity or causes pain, the surgical options include PKP or Descemet-stripping endothelial keratoplasty (DSEK).7,30,31,32,33 Although the rate of graft failure is high with need for subsequent corneal and glaucoma surgery, clear grafts with improved vision can be achieved. Controlling the preoperative and postoperative IOP is imperative in decreasing the risk for rejection after surgical intervention.7,30,31
Posterior Polymorphous Dystrophy
Posterior polymorphous dystrophy (PPMD) is an uncommon, typically nonprogressive, hereditary endothelial corneal dystrophy first described by Koeppe in 1916.34 Although the condition is bilateral, involvement may be so asymmetric as to be clinically evident in only one eye.35,36 PPMD is a genetically heterogeneous disorder inherited in an autosomal dominant fashion with incomplete penetrance; mutations associated with this disorder have been localized to chromosomes 1, 10, and 20.37,38,39 Mutations in the TCF8 transcription factor gene on chromosome 10 have been implicated in half of the cases of PPMD.40 In most patients, only limited portions of Descemet’s membrane and the corneal endothelium are involved.35,36 These patients are usually asymptomatic and may be unaware of their condition. In one series, fewer than 10% of the 120 patients with PPMD progressed to significant corneal edema and decompensation necessitating PKP, and fewer than 15% of patients developed ocular hypertension.36
Clinical Features of PPMD
PPMD can be categorized into three forms— vesicular, band, and diffuse—on the basis of the clinical appearance on slitlamp examination41 (Fig. 54.3). Approximately 40% to 50% of eyes with PPMD have vesicular or band lesions, and roughly 10% of eyes with PPMD have diffuse disease.39 Because PPMD can clinically resemble the ICE syndrome on slitlamp examination, endothelial specular photomicroscopy can be useful in accurately diagnosing PPMD.34,41 Using endothelial specular photomicroscopy, PPMD appears on the endothelium as an oval dark ring surrounding a lighter, mottled central area with scalloped edges.34 In patients with more-severe PPMD that has progressed to diffuse corneal edema, evaluation by slitlamp and specular microscopy becomes limited and in vivo confocal microscopy may be more useful in establishing the diagnosis.37,39,42,43 Using confocal microscopy, the endothelial abnormalities seen in more severe disease include multilayered endothelial cells and some of the endothelial cells take on morphologic characteristics of epithelial cells.37,44,45
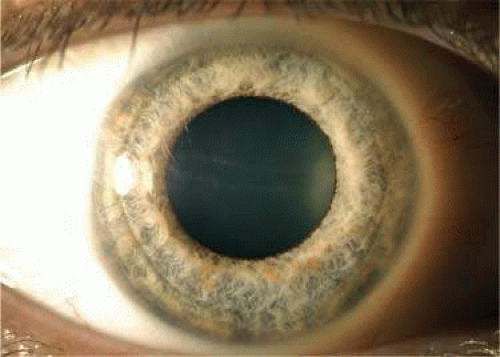 Figure 54.3. Slitlamp photograph of an eye of a patient with posterior polymorphous corneal dystrophy. There is a subtle band of “snail tracking” characteristic of this condition crossing the pupillary axis horizontally. |
Pathophysiology
The prevailing theory on PPMD is that it is an inherited disorder of the posterior nonbanded portion of Descemet’s membrane, causing an anomalous corneal endothelium that is present at birth.44 The appearance of epithelial-type cells amid the PPMD vesicles and bands may indicate that a common reaction of a distressed endothelial cell is to undergo metaplastic transformation to an epithelial phenotype; alternatively, it may be specific to PPMD.44,45 The abnormal endothelial cells, with or without epithelial characteristics, have been found to extend across the trabecular meshwork and onto the surface of the iris, obstructing the angle.36,46 These adhesions vary from PAS to prominent iridocorneal adhesions. In some cases, translucent membranes, apparently extending from the posterior cornea, are visible on the surface of the iris; these membranes can occasionally cause ectropion uvea and corectopia.36,47
Posterior Polymorphous Corneal Dystrophy and Glaucoma
Krachmer36 reported that 17 of 120 patients (14%) examined with PPMD had elevated IOP. Because so many patients with PPMD are asymptomatic, the true incidence of elevated IOP with this condition is probably much lower. Most of the patients in Krachmer’s group with elevated IOP had iridocorneal adhesions that closed portions of the filtration angle, resulting in a secondary angle-closure form of glaucoma. A second group of patients with PPMD and elevated IOP who have open angles but no iridocorneal adhesions has been recognized.35,36 The cause of the pressure increase in these patients is uncertain; in one such case examined histopathologically, an abnormally high insertion of the iris into the posterior trabecular meshwork, with some collapse of the intertrabecular spaces, was noted.48
Management
When present, glaucoma is best managed initially by using medications that decrease aqueous production. In those patients whose IOP cannot be controlled medically, laser trabeculoplasty is usually ineffective because the angle is often obstructed by membrane-like material or synechiae. When patients with PPMD develop glaucoma that is refractory to medical therapy, incisional glaucoma surgery—trabeculectomy with antimetabolites or GDD implantation—is often necessary. Insufficient research has been done to evaluate whether trabeculectomy or GDD implantation provides for better management of secondary glaucoma associated with PPMD.
In patients with significant corneal edema who require PKP, preoperative glaucoma was found to be a poor predictor of long-term keratoplasty success. In Krachmer’s series,36 among 13 grafts done on 7 patients with PPMD who had preoperative IOP elevation, 9 eyes (69%) had final visual acuities of 20/400 (6/120) or less after 2 to 9 years of follow-up; in comparison, all 9 eyes without preoperative IOP elevation had visual acuities better than 20/400 after transplantation. Although the numbers in this series are small, these findings suggest that the presence of preoperative pressure elevation is a poor prognostic factor for keratoplasty success in patients with PPMD.36
Fuchs Endothelial Dystrophy
In 1910, Fuchs described a hereditary bilateral condition of the cornea that included central corneal clouding with epithelial edema. This process is thought to occur secondary to a diseased endothelial cell layer of the cornea. This degenerative condition is bilateral but often somewhat asymmetric, and Fuchs postulated that the dystrophy affected fewer than 1% of his eye clinic patients.49 Fuchs endothelial dystrophy (FED) is probably an autosomal dominant disorder with incomplete penetrance; women older than 40 years of age are three times as likely to be affected as men in the same age group.50 Whether FED is caused by an inherited or acquired defect in the mitochondrial DNA is unknown.50
Clinical Features
In the early stages of FED, the patient is often asymptomatic. Symptoms begin when the cornea becomes edematous, and patients develop painless decreased vision and glare that are worse on awakening.49 On slitlamp biomicroscopy, guttae can be visualized on the corneal endothelium. (Fig. 54.4). When epithelial and subepithelial bullae form and rupture, pressure is placed on the corneal nerve endings, causing severe pain.50 In the end stage of the disease, scar tissue forms and the painful episodes become less frequent, but vision remains severely limited.49
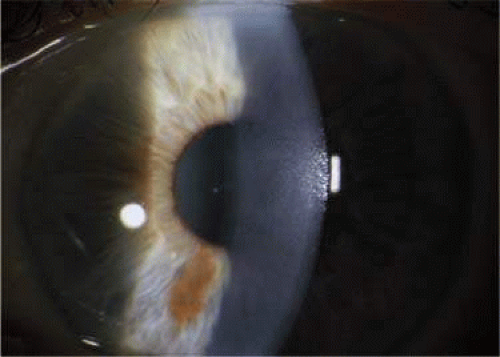 Figure 54.4. Slitlamp photograph of the cornea of a patient with Fuchs endothelial dystrophy. Fine guttata can be visualized on the surface of the corneal endothelium. |
Histopathology
Using light microscopy, patients with FED exhibit thickening of the posterior nonbanded layer of Descemet membrane along with the presence of guttae or wartlike excrescences on the posterior surface.49,51 The guttae look like hyporeflective areas surrounded by hyperreflective endothelial cells on specular microscopy, which also reveals that the endothelial cells are reduced in number, increased in size (polymegathism), and have a variable cell shape (pleomorphism), corresponding to the diseased endothelial cells that have replaced the normal corneal endothelium that is seen on scanning electron microscopy.51 Scanning electron micrographs of the endothelial cell layer have shown endothelial cells that have transformed into fibroblasts, epithelial-type cells, and degenerated fibroblast-like cells.49,51,52
Pathophysiology
Using immunohistochemical techniques, Hidayat and Cockerham52 showed that some cytokeratins normally restricted to the epithelium are present in the endothelium in FED. It has also been shown that corneas with FED have increased rates of apoptosis when compared with normal corneal endothelium.51 It is currently unknown whether this endothelial metaplasia—and perhaps additionally the increased rate of apoptosis—represents a nonspecific response of distressed endothelial cells, as previously reported with PPMD and ICE syndrome, or if it is part of the pathophysiology of the dystrophy. The dysfunctional endothelium may be caused by a hereditary defect in the function or expression of the Na-K ATP-ase pump, which is responsible for active transport of electrolytes and fluid out of the stroma and into the aqueous humor to maintain corneal dehydration.51 Another proposed theory is that the abnormal corneal endothelium might be caused by a hereditary defect in the terminal differentiation of the neural crest cells, from which the corneal endothelium is derived.49
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


