CHAPTER 2 Fundamentals of Molecular Biology and Gene Therapy
Fundamentals of Molecular Biology
Gene Expression
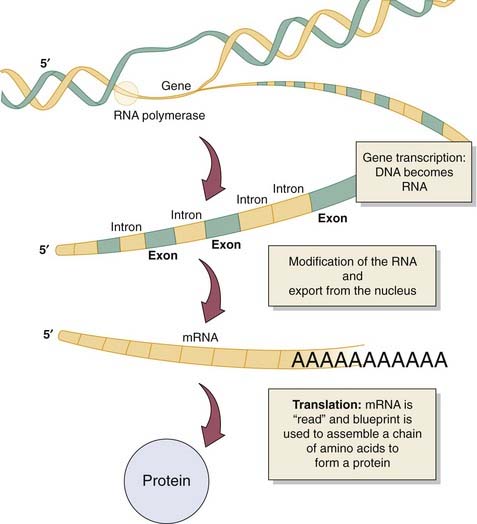
The process by which a gene codes for a specific protein begins with transcription, which is the formation of a single-stranded RNA molecule, which complements a single strand of the DNA subunit (Fig. 2-1). This RNA molecule is subsequently modified to become the messenger, which brings the genetic information or directions from the nucleus to the cell’s cytoplasm where the actual synthesis of the functional protein occurs. After reaching the cytoplasm, the process of translation begins (see Fig. 2-1). During translation, the message from the RNA molecule directs the construction of a protein from its basic subunit, the amino acid. Once a protein is formed, it may require further modification or control steps to enable its designated function. These modifications include the addition of sugars, lipids, or phosphates to the protein backbone. These internal control steps are mediated by existing cytoplasmic enzymes, which, if defective, can also lead to defined diseases. Once a protein is formed within the cell cytoplasm, it can reside in the cell or be released to affect other tissues within the body. Depending on the original genetic program for the protein, it may serve its purpose and be rapidly degraded, or it may enter the circulation or reside in local or distant tissues for an extended period. Although stated simply here, both transcription and translation are complex processes that have many more modification and regulatory steps governed by regulatory genes and their protein products. When these delicate control mechanisms are lost, a disease state or a state of abnormal cell proliferation can ensue, as is exemplified in the development of cancer.
Cell Division
Disorders in cell function can lead either to cell death or proliferation. Abnormal stimulation of cell proliferation is the basis for the development of cancer. For a cell to divide, it must progress through the various phases of growth and reproduction that constitute what is called the cell cycle (Fig. 2-2). The principal components of the cell cycle are the replication nuclear DNA and its distribution among the progeny cells (i.e., daughter cells). The first phase of the cell cycle is called G1, and it is here that all the enzymes, nucleic acids, and other factors are produced that will enable DNA replication or “synthesis,” which occurs in the S phase. Once the DNA is replicated, a period of cell growth and duplication of cellular proteins and structures occurs in a period called G2. After this cell growth, the actual distribution of the replicated DNA and the physical division of the parent cell into two daughter cells occur in the M phase, which takes approximately 1 hour. After cell division, the cell cycle process can begin again, or the cell may enter a state of rest called G0.
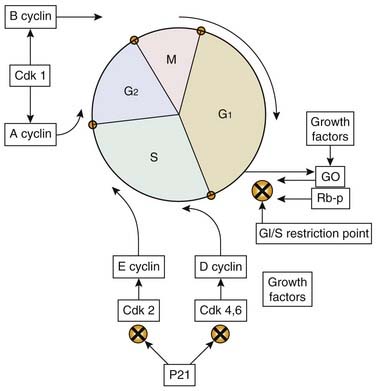
Continued molecular research will provide further understanding of cell cycle regulation and may lead to novel therapies for treating human cancer. A discussion of basic molecular biology techniques, which have provided the means to understand gene expression, regulation, and cell cycling, is found in Chapter 16.
Gene Therapy
Replacing Defective Genes
During the advent of gene therapy, the initial clinical targets were rare inherited diseases such as adenosine deaminase deficiency, which results in a deadly systemic immune deficiency, enzymatic deficiencies resulting in cystic fibrosis and liver disease, or coagulation pathway deficiencies resulting in various types of hemophilia. Gene therapy could be used to introduce a normal gene into the body to compensate for the low or absent function of the patient’s mutant gene. For example, a proportion of children born with severe combined immunodeficiency (SCID) have a deficiency in adenosine deaminase (ADA) due to a mutation in the gene encoding this enzyme. Several clinical trials have introduced a functioning ADA gene into the bone marrow of SCID children using a viral vector, effectively reconstituting the patient’s immune system.1 Clinical trials assessing the potential for the treatment of hemophilia B through the transfer of a functioning factor IX gene, and cystic fibrosis via introduction of an intact cystic fibrosis transmembrane conductance regulator (CFTR) gene have also been completed.2,3 These studies strongly suggest future applicability for gene therapy in the treatment of inherited diseases.
Enhancing Gene Expression
In the past decade, it has become clear that gene therapy may have its most immediate and effective role in treating more common acquired diseases, including various cancers, arthritis, and atherosclerosis.4,5 In order to tackle these conditions, new approaches have been adopted that focus on increasing the expression of beneficial genes beyond normal levels. For example, in a clinical trial for the treatment of melanoma, tumor-infiltrating lymphocytes (TILs) were purified from a tumor at the time of surgical resection.6 The TIL cells were then grown in the laboratory, and the gene for tumor necrosis factor (TNF) was introduced into the cells.7 The genetically engineered cells were subsequently transfused into the patient and preferentially migrated to the site of residual tumor, delivering a therapeutic dose of TNF. As the molecular basis for disease is elucidated, there is likely to be increasing interest in this form of gene therapy.
Suppressing Gene Expression
Most recently, as the molecular pathways that underpin disease—particularly cancers—are unraveled, there has been excitement about the possibility of suppressing key genes known to be involved in the disease process. For example, RET, a proto-oncogene implicated in the pathogenesis of medullary thyroid carcinoma, can be inactivated by inducing expression of a mutant form of the RET gene, resulting in tumor regression.8 This approach is dependent on the “dominant negative” effect where the mutant gene encodes a protein that retains binding ability to other key proteins that normally interact with the wild-type gene. However, the mutant protein is nonfunctional, creating a competitive inhibition of the wild-type functional variant. Dominant negative therapies are presently being translated to clinical trials in the hope of developing novel treatments, particularly within the field of oncology.
A new technique that may have immense potential for the future of gene therapy is RNA interference (RNAi), a method by which genes can be selectively “silenced.” RNAi is a ubiquitous mechanism that is used by the cell to effectively “turn off” specific genes, as described a decade ago in a landmark paper.9 In this study, injection of double-stranded RNA consisting of both sense and antisense strands into the nematode Caenorhabditis elegans profoundly silenced the gene with a sequence complementary to the dsRNA. The potential for RNAi in research and therapeutics was immediately recognized. In effect, any gene, including those involved in cancer, could be silenced and the effect of this loss of function on phenotype could be elucidated. The scientists who discovered RNAi were awarded the Nobel Prize in Medicine in 2006, only 8 years after publication of their initial, landmark paper.
Subsequent studies have partially determined the mechanism for RNAi (Fig. 2-3). In brief, an RNAase enzyme called Dicer cleaves dsRNA to form 21- to 23-nucleotide fragments of RNA. This short-interfering RNA (siRNA) is the actual effector molecule in RNAi.10–12 The siRNA is integrated into the RNA-induced silencing complex (RISC), after which the sense strand of the duplex is discarded. The antisense siRNA is retained and acts as the “guide strand,” directing RISC to mRNA molecules with a complementary sequence.13 RISC cleaves the targeted mRNA, thereby silencing gene expression by blocking the translation of the mRNA sequence to protein.10,14
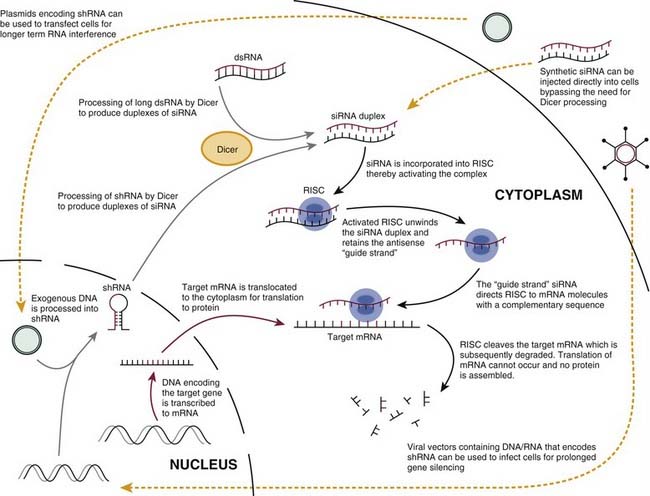
Considerable progress has been made in applying RNAi to mammalian systems. Transfection of mammalian cells with long dsRNA molecules of more than 30 nucleotides was found to induce a significant host antiviral interferon response that produced a general silencing of untargeted genes and often cell death. This hurdle has recently been overcome by chemically synthesizing 21-nucleotide siRNA molecules and using these to induce efficient, targeted gene silencing in mammalian cells without inducing an interferon response.11 These siRNAs can be used in subnanomolar concentrations to achieve greater than 90% reduction in mRNA levels, a highly potent gene-silencing effect.10 Recent animal experiments involving siRNAs have indicated that immune system activation is relatively uncommon, bolstering the safety profile of these novel therapies.
Unfortunately, a persisting problem associated with RNAi techniques are “off-target” effects. This refers to unintended siRNA-mediated silencing of genes that have similar nucleic acid sequences. Even small amounts of sequence homology, as few as seven base pairs, are enough to produce off-target effects. Several computer algorithms have been designed to aid in the selection of siRNAs with minimal off-target effects. It is unclear what effect, if any, off-target effects will have in human trials. In fact, early human trials of RNAi therapies for the treatment of macular degeneration and neurodegenerative diseases have not revealed any significant side effects.12
Somatic versus Germ Cell Gene Therapy
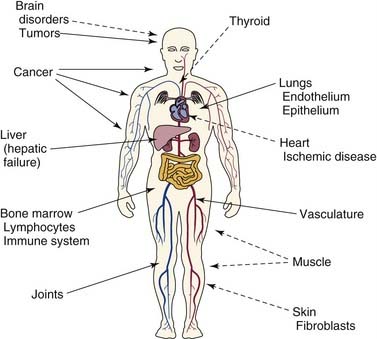
Gene therapy has two possible target cell types. The first and presently used targets are the somatic cells, or those cells that constitute the organs and postnatal tissues of the body. The second potential target is the germ cells, or those cells that produce the sperm or ovum and are passed on to a person’s offspring. Many different organs and cell types are targets for somatic gene therapy, including bone marrow, liver, tumor cells, muscle, skin, endothelium, thyroid, and others (Fig. 2-4). Genetic manipulation and therapy at these sites does not alter the inherited genetic material and raises few novel ethical or social issues.15 Genetic manipulation of the sperm or ovum, however, could prevent inherited diseases by altering the genetic constitution of offspring. Although this is an appealing idea on the surface, there are serious technical and safety issues as well as profound ethical concerns involved. Currently, gene therapy is restricted to somatic cells, and genetic manipulation of human germ cells would be prohibited under existing recombinant DNA guidelines.16
Methods of Delivering Gene Therapy
Direct Injection of Genetic Material
DNA-Mediated Transfer of Therapeutic Genes
The process of DNA-mediated gene transfer is called transfection, and the vehicle through which genetic material is transferred into a cell is called a vector. Functional DNA vectors are circular molecules of DNA called plasmids that contain various additional genetic elements required to achieve expression of the gene product at therapeutic levels (Fig. 2-5). Included in these are the special elements (promoter and enhancer) that direct gene expression and elements that determine the processing and persistence of genetic material within the cell. Plasmid vectors can be used to effectively deliver therapeutic genes to target cells. Plasmids have also been used to prolong the transient, 3- to 7-day gene-silencing effect associated with the transfection of naked siRNAs.
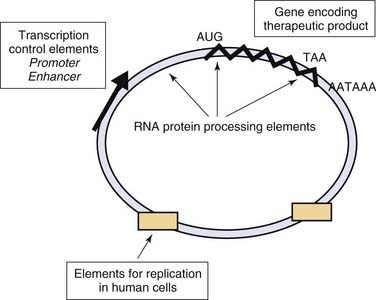
The delivery of DNA vectors into cells is possible via a variety of techniques. One classic method is to simply microinject plasmids directly into the cell nucleus.17 This technique is both time-consuming and inefficient for achieving large numbers of transfected cells. Although this method is common in the laboratory with in vitro studies, its technical limitations prohibit effective application to living animal models or human subjects. A more efficient process, which also is limited to in vitro application, is the process of electroporation, in which cultured cells are exposed to DNA in the presence of a strong electrical pulse.18 The electrical pulse creates pores in the cell membrane, which allows electrophoresis of plasmid DNA into the cell.
It is possible to effectively introduce genes into muscle19 or thyroid20 simply by injecting DNA into these tissues in vivo, where the process of endocytosis enables cellular uptake. Gene transfer into other organs requires special methods to enhance the uptake of DNA into the specific target cells. A common alternative method is the use of cationic lipids, which encase DNA vectors and fuse with the target cell membrane21 to enhance intracellular gene uptake. This process has been termed lipofection. Another method is to couple the DNA vector to proteins that bind to specific receptors on the target cell leading to uptake of the DNA by receptor-mediated endocytosis.22,23 A novel approach to DNA vector delivery involves the use of the “gene gun,” which uses electrical currents to project microscopic gold beads coated with plasmid DNA into target tissues and organs.24
An important point to understand is that DNA-mediated gene transfer typically results in only transient residence of the therapeutic genes in the targeted cell. DNA vectors, which are introduced into cells, are degraded and eliminated from the cell over time. Different cell types eliminate the introduced genetic material at different rates. In muscle, for example, DNA vectors may persist in cells for many months and continue to express gene products.19 In contrast, DNA vectors injected into the thyroid have a shorter half-life and the gene product is eliminated after 2 days.20 Vectors introduced into the liver are eliminated with a half-life of approximately 1 to 2 hours and expression is significantly reduced after 6 to 24 hours.25
Direct Delivery of RNA for Gene Silencing
RNA can also be introduced into cells, opening the door to potential therapeutic implementation of gene silencing. Intravenous administration of siRNA targeting the VEGF gene, for example, has been shown to significantly reduce tumor volume and intratumoral VEGF levels.26 Unfortunately, the gene-silencing effect of synthetic siRNAs lasts only minutes to a few days before they are degraded. Chemical modification of the RNA can improve stability. The plasma half-life of an unmodified siRNA in a mouse model was 0.03 hour, the half-life of a chemically modified siRNA was 0.8 hour, and the half-life of a lipid-conjugated, modified duplex was 6.5 hours.27
Despite the improved stability of modified siRNA, plasma clearance is still rapid and any potential therapeutic benefit will require using chemically modified RNA in combination with some kind of delivery system.10 Liposomes have been successfully used to deliver siRNA in vivo, and significantly improve both the duration and level of gene suppression compared to nonvector delivery methods. More recently, nanotechnology has been used to design compounds consisting of a cationic polymer that binds the RNA cargo, a neutral “stealth” coating to hide the particle from immune surveillance and a targeting ligand. This type of nanoparticle has been used to selectively target neuroblastoma tumors in nude mice with anti-VEGF receptor siRNA, resulting in a dramatic antitumor effect.28 Electroporation and “gene gun” techniques have also been used to effectively deliver siRNA in vivo.10
The most common method used to date for the delivery of siRNA is termed hydrodynamic delivery. This approach involves injecting a large volume of an RNA-containing solution as a rapid intravenous bolus. The hydrostatic pressure generated transiently disrupts the vascular endothelium and achieves siRNA delivery to tissues. To date, hydrodynamic delivery has been successfully used to deliver siRNA to skeletal muscle in rats, dogs, and rhesus macaques.29 Localized delivery can be achieved by application of a tourniquet to isolate a limb before intravenous injection. The potential for this approach in humans is under investigation and will likely transition to clinical trials soon.10
The need for sustained gene silencing has led to the development of novel DNA plasmid expression vectors that direct the synthesis of short RNA over a prolonged period (see Fig. 2-3). The encoded short RNA sequence is transcribed and processed into the effector gene-silencing RNA molecule. One method involves genetically engineering a plasmid to encode a short-hairpin RNA (shRNA). A shRNA molecule consists of a special double-stranded RNA molecule that includes a sharp hairpin turn and is processed in the cell to yield a 21-nucleotide RNA construct capable of gene silencing. Use of DNA plasmid vectors can prolong in vivo mammalian gene silencing for up to several weeks.12 They are, however, limited by their inability to transfect nondividing cells.11 This limitation can be overcome, however, through the use of viral vectors to deliver siRNA, which is discussed in the next section.
Viral-Mediated Gene Transfer
Retroviruses
The original prototypes for viral-mediated gene transfer are retroviral vectors derived from the Moloney murine leukemia virus.30,31 Retroviral vectors were chosen as vehicles because of several useful properties. First, “defective” virus particles can be constructed that contain therapeutic genes and are capable of infecting cells, but which contain no viral genes and express no pathogenic viral gene products. A general scheme for constructing a defective retroviral particle is illustrated in Figure 2-6. Second, retroviral vectors are capable of permanently integrating the therapeutic genes they carry into the chromosomes of the target cell. Because of this property, retroviral vectors are well suited for treating diseases that require permanent gene expression. Third, modifications can be made in retroviral vectors and in the cell lines producing vectors that result in enhanced safety features.
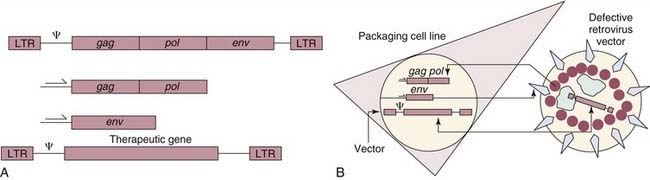
Previous experience in animal models32,33 and initial experiences in clinical trials suggest that these vectors are, generally, safe. However, there have been serious complications in clinical trials using retroviral vectors to treat X-linked severe combined immunodeficiency. Although gene therapy reconstituted the immune system and produced excellent clinical outcomes in most children, several cases of T-cell leukemia were reported 2.5 to 5 years after therapy.34 This has been directly attributed to the insertional oncogenesis that can occur when the retroviral vector permanently inserts into the host genome and activates nearby cellular proto-oncogenes. The powerful promoters used to drive expression of the therapeutic gene may also activate nearby genes.35 Consequently, newer generation retroviral vectors are in development that use weaker promoters to minimize trans-activation of genes. Special “insulator” sequences can also be used to block the transcriptional elements of the vector from interacting with cellular genes. Another option is to include “suicide” genes in the vector that will initiate cell death in overproliferating cells.36 These methods have shown early promise but have yet to be used in a clinical trial.
Adenoviruses
A recent focus of gene therapy has been the development of adenovirus vectors as powerful and effective vehicles for gene transfer.37 An overview for the construction of a replication-defective adenoviral vector is shown in Figure 2-7. Adenoviral vectors differ from retroviral vectors in that they remain episomal; that is, they do not integrate their genes into the target cell’s chromosome. Compared with retroviral vectors, adenoviral vectors demonstrate the significant advantage of infecting a wide variety of both dividing and nondividing cells in vitro and in vivo with a high level of efficiency.38–40 Using adenoviral gene transfer, expression of the therapeutic gene is possible for a period of several weeks to months. More recently, adenoviral vectors have been designed to encode and transiently express shRNA, which may be particularly useful in therapeutic strategies against cancers where long-term RNAi is not necessarily desirable.12
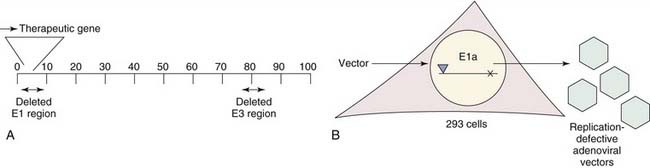
First-generation adenoviral vectors contained a deletion of portions of their E1a and E3 regions with the former acting as a “master switch” regulating the expression of other viral genes that are critical for viral replication. Consequently, all first-generation adenoviral vectors are replication-deficient. However, despite these modifications, several of the remaining viral genes are still expressed, markedly increasing immunogenicity.4
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


