Chapter 48 Flecked retina disorders
Introduction
“Flecked retina” describes disorders characterized by multiple yellow-white retinal lesions of various size and configuration.1,2 A number of inherited or acquired conditions, associated with a variable degree of retinal dysfunction, correspond to this definition. This chapter reviews the differential diagnosis of flecked retina in childhood; some disorders predominantly seen in adults are also included.
Clinical evaluation
It is important to enquire about visual symptoms such as blurred vision, nyctalopia, or hemeralopia and obtain a full family history. Examination of relatives can be helpful, as other affected family members may remain asymptomatic. Current or previous systemic drug administration and dietary habits should be enquired after. Careful note should be taken of any other medical disorders particularly those associated with malabsorption (e.g. cystic fibrosis).3
Yellow-white retinal lesions in primary ocular disease
Stargardt’s disease and fundus flavimaculatus
Stargardt’s disease is a typically autosomal recessive retinal dystrophy, associated with mutations in the ABCA4 gene.4 Pseudo-dominant inheritance where an affected parent (whose partner is unknowingly a carrier of the disorder) may have an affected child can be observed particularly in consanguineous families.5 In such cases, it is important for counseling to distinguish ABCA4-related disease from phenotypically similar autosomal dominant disorders due to mutations in ELOVL46 or RDS.7 Stargardt’s disease is characterized by the presence of yellow-white flecks that are scattered throughout the posterior pole (Fig. 48.1). Macular atrophy is common and affected individuals typically present with reduced visual acuity in the second decade of life. Some patients have good central vision with minimal or no macular involvement at presentation, and may be diagnosed as fundus flavimaculatus.8 Stargardt’s disease and fundus flavimaculatus are parts of the phenotypic spectrum of the same disorder and the same genetic etiology is frequently observed. Patients rarely complain at presentation of poor night vision or field constriction although rod function can be abnormal.9–10
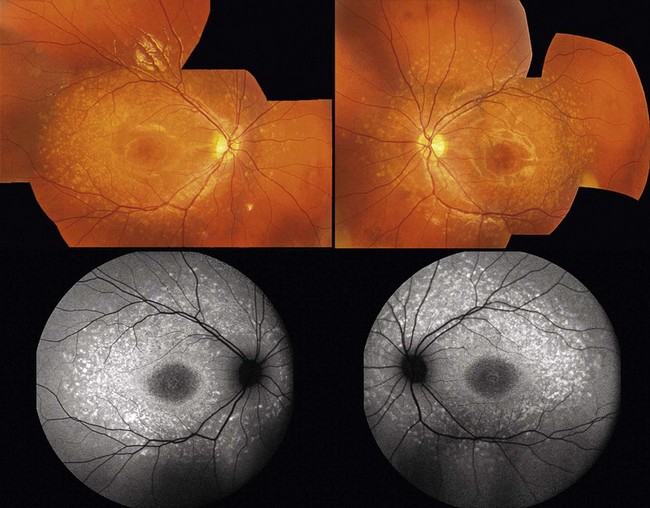
FAF is very useful in diagnosing individuals with Stargardt’s disease; it allows detection of the abnormal phenotype even in cases when it is not otherwise evident. Abnormalities in the form of a speckled hyper- and hypo-autofluorescence pattern and/or focal areas of increased signal, spatially correlating with flecks on fundoscopy, can be identified even in the early stages.11 SD-OCT, in particular the degree of preservation of the line corresponding to the inner/outer segment junction, allows more accurate assessment of disease severity. A “dark choroid” has been classically described on fluorescein angiography, although this is now not a commonly used diagnostic tool for Stargardt’s disease, particularly in children.12
Electrophysiologic testing reveals an abnormal pattern ERG and attenuated central macular responses on the multifocal ERG. The results of full-field electroretinography are variable and often normal in the early stages of disease. Full-field ERGs may be prognostically useful since individuals with early evidence of rod and/or cone photoreceptor dysfunction are at more risk of severe and progressive disease.9
Benign fleck retina
Benign fleck retina is an autosomal recessive condition associated with a distinctive retinal appearance and no apparent visual or electrophysiologic deficits.13 Affected individuals are asymptomatic, but fundus examination reveals a striking pattern of diffuse, yellow-white flecks extending to the far periphery, sparing the foveal region (Fig. 48.2).14–16 The flecks are located at the level of the retinal pigment epithelium (RPE) and can be present in early infancy.
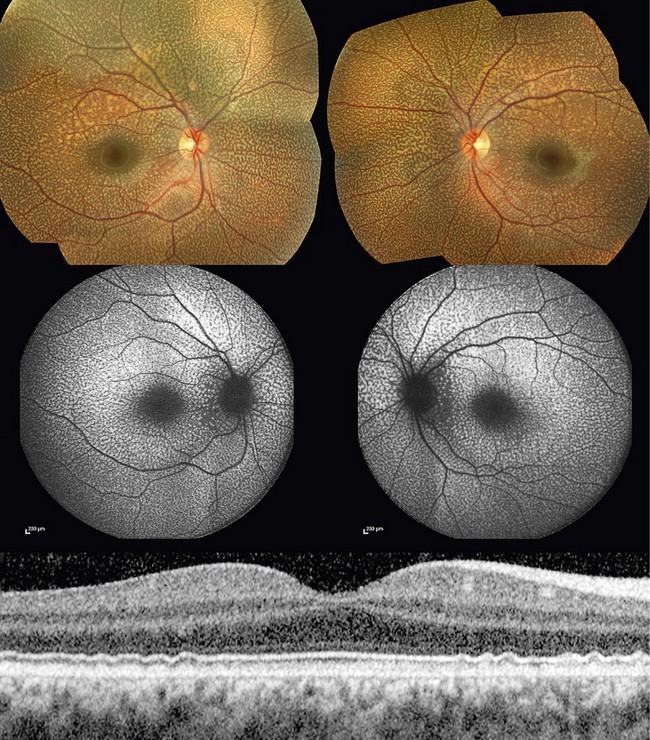
FAF reveals multiple hyperautofluorescent lesions corresponding in location with the flecks. On SD-OCT, discrete deposit accumulation located posterior to the photoreceptor inner/outer segment junction but without disrupting it is observed. Normal full-field and pattern ERGs confirm the diagnosis;15 multifocal ERG may be slightly subnormal in some isopters.16
Recessive mutations in the gene encoding group V phospholipase A2 (PLA2G5) and mildly elevated low-density lipoprotein and total cholesterol levels have been identified in some individuals with benign fleck retina.17
Benign fleck retina should not be confused with “fleck retina of Kandori.” The latter is associated with large white lesions, possibly atrophic changes, and night-blindness.18 It is not clear if “fleck retina of Kandori” is a genetic condition or even an independent clinical entity.
Fundus albipunctatus
Fundus albipunctatus is an autosomal recessive condition associated with night-blindness and numerous widespread yellow-white punctate lesions at the level of the RPE (Fig. 48.3).19–20 Patients are either noted to have an abnormal retinal appearance on a routine eye test or present with nyctalopia. Affected individuals describe night-blindness from birth and delay in dark adaptation after exposure to bright light.19 Visual acuities and visual fields are usually normal.
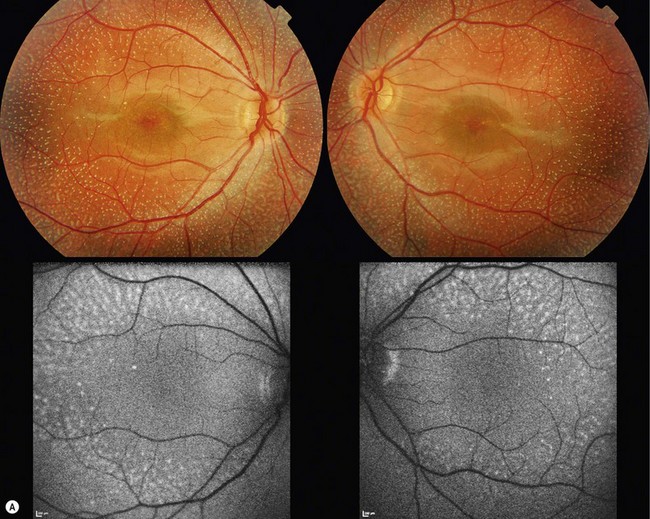
FAF in individuals with fundus albipunctatus often reveals a low autofluorescent signal; digital enhancement artefacts such as autofluorescence from the optic disk and large vessels may be observed.21 In young subjects, high-density foci partially associating with the white dots on fundoscopy can be observed (Fig. 48.3A).19 Hyper reflective lesions at the level of the RPE are detected on SD-OCT (Fig. 48.3B).19
Characteristic abnormalities on electrophysiology and dark adaptometry facilitate the diagnosis (see Chapter 44). Notably, dark-adapted ERG responses are subnormal following conventional dark adaptation but partially recover or normalize following extended dark adaptation.19,22 Light-adapted ERGs and pattern ERGs can be normal or abnormal.19,23,24
Molecular testing can be useful: fundus albipunctatus is commonly associated with recessive mutations in RDH5, a gene encoding an enzyme with 11-cis-retinol dehydrogenase activity.25 This enzyme catalyzes the oxidation of 11-cis-retinol to 11-cis-retinaldehyde, the universal vertebrate chromophore of visual pigments, in human RPE.26–27 Mutations in the RPE6528 and RLBP129 genes have also been described in patients with some features of fundus albipunctatus.
Retinitis punctata albescens, Bothnia and Newfoundland retinal dystrophies
Retinitits punctata albescens is a variant of retinitis pigmentosa characterized by multiple retinal white dots rather than pigment deposition.30 Patients have family history consistent with autosomal recessive inheritance and present with nyctalopia and/or peripheral vision problems; decreased visual acuity may also occur.31 On fundoscopy, white spots similar to the ones observed in fundus albipunctatus are observed (Fig. 48.4). These spots spare the foveal region and may evolve to give rise to a more classical pigmentary retinopathy. It is important to differentiate early retinitis punctata albescens from fundus albipunctatus: the prognosis for daytime vision is considerably better in the latter and the former features more severe and progressive retinal degeneration. Vascular attenuation, visual field loss, and a more severe electrophysiologic phenotype are key distinguishing features (see Chapter 44).31
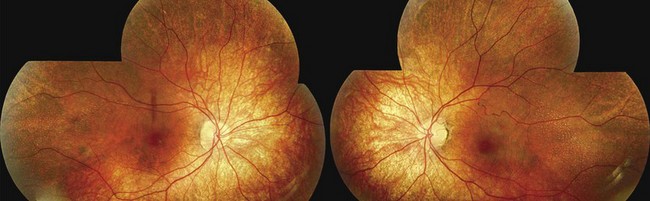
Retinitis punctata albescens is primarily associated with recessive mutations in the cellular retinal binding protein-1 gene (RLBP1; see Chapter 44).32 Mutations in the RDS33 and the rhodopsin34 genes have also been described in rare cases of dominantly inherited retinitis punctata albescens.
Newfoundland rod–cone dystrophy35 and Bothnia dystrophy36 are two early-onset forms of retinal disease that have high prevalence in the genetic isolate populations of northeastern Canada and northern Sweden, respectively. Both share genetic etiology (RLBP1 mutations) and key phenotypic features (multiple white dots, night-blindness) with recessive retinitis punctata albescens35,37,38 and are parts of the phenotypic spectrum of the same disorder.
Enhanced S-cone syndrome and Goldmann-Favre syndrome
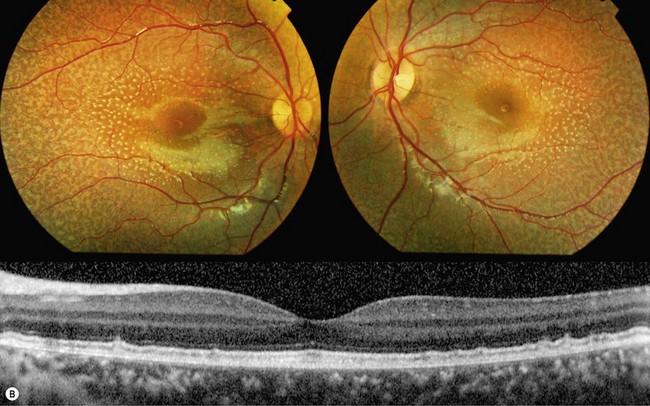
Small subretinal white dots can be an early feature of a progressive retinopathy named “enhanced S-cone syndrome” (ESCS).39–42 Patients typically present with night-blindness and/or visual acuity loss in the first or second decade of life; a hyperopic refractive error is often noted.40 ESCS has been associated with various fundoscopic features: the most typical are retinoschisis and deep nummular pigmentary deposition with or without yellow-white spots, primarily along the vascular arcades.43,44 White dots have been described in pediatric patients (Fig. 48.5); those tend to progress to a pigmentary retinopathy in later adulthood.41,42,45
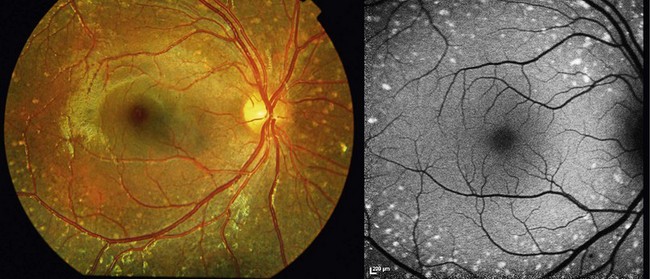
FAF reveals abnormal signal, in particular beyond the vascular arcades; mid-peripheral hyperautofluorescent spots only partially corresponding to fleck-like abnormalities on fundoscopy are often observed in young subjects.42,44 A hyperautofluorescent ring has also been described.40 Cystic spaces, rosette-like lesions, and/or schisis may be observed on OCT.40,42–44
ESCS is unique among retinal dystrophies: there is both retinal degeneration resulting in visual loss and an increased function in a subset of photoreceptors, the S-cones.46–49 A distinctive electophysiologic phenotype is observed.40
ESCS is associated with recessive mutations in the NR2E3 gene, encoding a retinal nuclear receptor with a role in activating rod development and repressing cone development.49,50
Goldmann-Favre syndrome was initially described as an autosomal recessive vitreoretinopathy characterized by night-blindness, pigmentary degeneration, macular and peripheral retinoschisis, cataract, and degenerative vitreous changes.51,52 Many of these features are shared with ESCS and appropriate electrophysiologic and genetic testing revealed that they are not distinct entities but two identifiable phenotypes in a wide spectrum of clinical expression of the same retinal degeneration.41,52,53
Miscellaneous reports of retinal flecks associated with inherited retinal disease
Small yellow-white fleck-like lesions have been reported in association with a number of other retinal disorders including early-onset retinal dystrophy due to RPE65 mutations54,55 and juvenile retinoschisis.56–58 In the former, the dots tend to be more fine and peripheral; in the latter, they are usually within the posterior pole. Additionally, autosomal recessive bestrophinopathy, a condition associated with biallelic BEST1 mutations, may present in childhood with small yellow-white subretinal deposits in the fovea and around the vascular arcades.59,60
Drusen-like deposits in inherited retinal disease
Drusen deposition at the macula is the hallmark of age-related maculopathy. Similar lesions may be seen in young adults affected with Sorsby’s fundus dystrophy (mutation in the TIMP3 gene61,62) or autosomal dominant drusen, also known as Doyne’s honeycomb retinal dystrophy63 or malattia leventinese64 (mutation in the EFEMP1 gene65) – two dominantly inherited maculopathies. Rarely, drusen-like lesions are observed in asymptomatic children as an incidental finding.1
Drusen-like deposits can be observed in children with North Carolina macular dystrophy. North Carolina macular dystrophy (see Chapter 46) is an autosomal dominant condition characterized by macular drusen that are present from birth.66,67 Fundoscopic findings are highly variable (Fig. 48.6) and tend to be more dramatic than one would predict from visual acuity measurements. Affected individuals may have:
1. Only a few drusen in the fovea (grade I)
2. Confluent drusen with or without RPE changes (grade II), or
3. Central macular chorioretinal atrophy with hypertrophic fibrous tissue (grade III).66,68
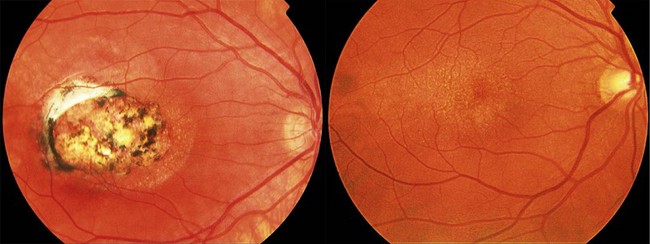
The condition rarely progresses; the grading does not reflect successive stages of progression.68–70 Visual acuity is stable and generally well-preserved except in those with central atrophic lesions. Development of choroidal neovascularization is uncommon but can result in fibrosis and late visual loss.66,70
Although the genetic defect is not known, the disease-causing gene has been mapped to the long arm of chromosome 6.70–72 A number of macular dystrophies that do not map to chromosome 6 but share features with North Carolina macular dystrophy (including macular drusen) have been described.73–75
Bietti’s crystalline corneoretinal dystrophy
Bietti’s crystalline dystrophy is an autosomal recessive disorder characterized by progressive chorioretinal degeneration, nyctalopia, reduced visual acuity, visual field constriction, and multiple yellow-white crystalline deposits at the retina, in circulating lymphocytes and, occasionally, at the corneoscleral limbus.76,77 Retinal crystals are observed predominantly at the posterior pole, in the superficial and deep retinal layers (Fig. 48.7). They are associated with multiple, sharply demarcated areas of RPE atrophy and tend to disappear as the disease advances.76,78,79 Affected individuals typically become symptomatic after the second decade of life.79,80 However, some pediatric cases have been described.81 Lesions partially corresponding to the crystals may or may not be evident on FAF imaging.82 SD-OCT reveals hyperreflective lesions throughout the retina; many of these lesions do not spatially associate with retinal crystals on fundus photography.82–84 When full-field ERG testing is performed, both cone and rod systems are found affected even in the early stages of disease.81,85,86
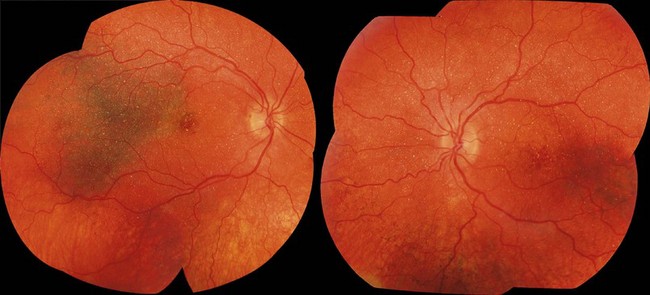
Bietti’s corneoretinal dystrophy is associated with defects in CYP4V2 (cytochrome P450, family 4, subfamily V, polypeptide 2),77 a gene encoding an enzyme with a role in fatty acid metabolism.87 A high heterozygous carrier frequency for CYP4V2 mutation, and therefore, relatively high prevalence of the condition, has been reported in East Asia, especially in Chinese and Japanese populations.80
A dominantly inherited retinopathy (with incomplete penetrance and variable expressivity) with clinical features similar to the ones observed in Bietti’s crystalline dystrophy has been described.88
Yellow-white retinal lesions in acquired disease
Vitamin A deficiency
Deficiency of the fat-soluble vitamin A (hypovitaminosis A) limits growth, weakens innate and acquired host defenses, and increases the risk of infectious morbidity.89–92 Ocular manifestations of hypovitaminosis A account for approximately 500 000 cases of childhood blindness worldwide usually as a result of corneal scarring (xerophthalmia).3,93 Vitamin A deficiency can be due to malnutrition (commonest cause of vitamin A deficiency in developing countries), malabsorption, or impaired metabolism of vitamin A associated with liver disease or cystic fibrosis.94
Chronic vitamin A deficiency may lead to visual symptoms as a result of reduced photoreceptor visual pigment formation.95 Night-blindness is a common complication and an early symptom of the disorder; visual field loss, abnormal color vision, and visual acuity loss may occur in later stages. Fundus examination often reveals multiple gray-white punctate spots at the level of the RPE and scattered in the peripheral retina.96–98 Atrophic and pigmentary changes may occur with disease progression. Dark adaptometry demonstrates elevated rod and cone thresholds, with rods more severely affected.99 Electrophysiology reveals reduced or undetectable rod-specific ERGs and reduced amplitude cone-flicker ERGs of normal implicit time.98,99
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


