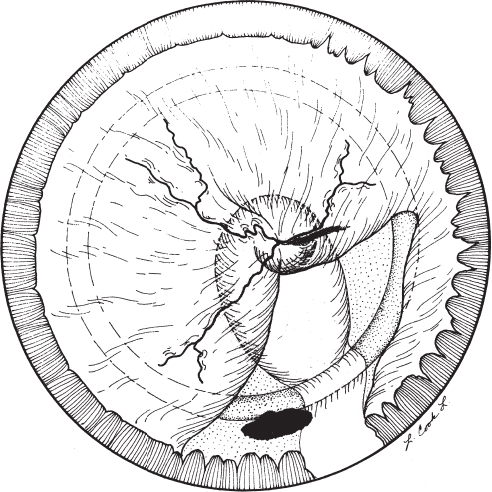
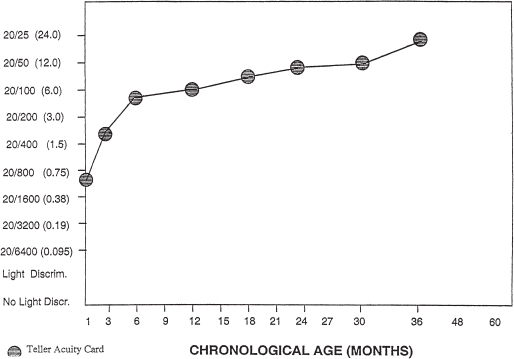
SECTION V In many retinal diseases, there is impaired central vision, visual field, night vision, or color vision. Pain is rarely present. In infants, testing for visual functions is challenging and may be difficult to interpret because the infant’s visual system is not fully developed at birth. For example, visual development of grating acuity measured with Teller Acuity Cards follows a curve where adult levels of normal acuity are not attained until after the age of 4 years1 (Fig. 20–1). Poor vision at birth may be missed because the newborn infant does not appear to respond to adult visual clues and does not describe visual symptoms. Therefore, in situations where the risk of poor vision exists, screening examinations of the retina are performed. As a child develops, certain milestones are noted. We describe vision by observing an infant’s behavior. At 6 weeks of age, a full-term infant should steadily fix on a large object, such as one’s face. By 2 months of age, the child will fix and follow large objects of high contrast. With further development, the child will reach for objects and be able to see smaller objects. The child should use both eyes together and not exhibit crossing of the eyes when attentively focusing on an object. Periodic crossing may be noted if the child is focusing on something up close or if he or she is fatigued and not attentively focusing, or it may be misdiagnosed if the child has broad epicanthal folds. If the parents are concerned, however, that there is eye crossing, and none is found on a single ophthalmologic examination, it is prudent to reexamine the child at another appointment. A young child with vitreoretinal disease may exhibit (1) failure to fix and follow an object at an appropriate age; (2) nystagmus; (3) amblyopia; (4) strabismus (Fig. 20–2); (5) conjunctival hyperemia associated with inflammation or elevated intraocular pressure; (6) difference in the size of the globes; or (7) an abnormality in pupillary reflex, such as leukocoria (Fig. 20–3). A child with vitreous floaters may describe these as familiar objects in his or her environment. For example, a child might say that he or she has a “stick” in his or her eye. In a child with a known retinal disease, parents should be suspicious of a possible problem if on more than one occasion a child describes an object in the visual space that is not present in reality. An infant may blink in response to a penlight. When a child is able to sit up, small interesting objects can be used to get the child’s attention. The examiner notes whether the child can maintain central and steady fixation with each eye. This can be done with a penlight to determine (1) whether the light reflex is in the center of the pupil, implying central fixation; (2) whether there is lack of ocular movement, implying steady fixation; and (3) whether the child maintains fixation with the eye of interest when the other eye is no longer occluded, implying maintained fixation. When the child’s pupillary reflexes are centered, the eyes are orthophoric. As the examiner moves an object or penlight, he or she notes whether the child moves his or her eyes to follow the object. The examiner then covers one eye at a time to see whether the infant still follows the object with each eye. If the child prefers one eye to the other, amblyopia is suspected. Nystagmus can be detected by determining if there is ocular movement while a child is fixating on a penlight or object. The nystagmus is described as monocular or binocular, and, if binocular, the oscillations are noted as asymmetric, conjugate, or disconjugate, meaning that the eyes will move in opposite directions. Disconjugate movement is described as horizontal or vertical and conjugate nystagmus is described as pendular or jerk.2 FIGURE 20–1. Visual development curve depicting visual norms obtained with Teller Acuity cards for infants aged 1 month to 4 years. (Data from Mayer DL, Beiser AS, Warner AF, Pratt EM, Raye KN, Lang JM. Monocular acuity norms for the teller acuity cards between ages one month and four years. Invest Ophthalmol Vis Sci. 1995;36:671–685, with permission). The triad of nystagmus, head bobbing, and abnormal head posture is called spasmus nutans. The nystagmus is often intermittent, of high frequency and small amplitude. It begins within the first year of life and lasts about a month to several years. It is often benign, but it is important to rule out other causes of monocular nystagmus or spasmus nutans, including tumors of the optic nerve, third ventricle, and thalamus, which occur in about 1% of cases.2 Bilateral nystagmus can be congenital, associated with spasmus nutans, or secondary to sensory deprivation. Nystagmus secondary to a sensory deprivation may not be noted at birth but develop soon after. It can be associated with reduced foveal reflexes a few months after birth when foveal maturation occurs (Fig. 20–4). Nystagmus may be associated with albinism, achromatopsia, optic-nerve hypoplasia, Leber’s congenital amaurosis, coloboma, aniridia, cone dystrophies, peroxisomal disorders, and central causes. When an infant has vision loss and nystagmus, the cause should be ascertained. Consultation with a neurologist and consideration of neuroimaging are recommended. Consultation with a geneticist may be indicated. Electrophysiology may be considered to rule out congenital stationary night blindness. In an older child, a secondary sensory nystagmus may be related to a retinal condition or multiple sclerosis. Infant and child grating visual acuity can be objectively determined by using preferential looking with Teller Acuity Cards3,4 especially in a child too young to measure with Snellen acuity. Preferential looking is based on one’s tendency to look at a patterned stimulus rather than a plain one. The stimuli used are equally spaced, highly contrasting stripes. By varying the width of the stripes or spatial frequency, the corresponding visual acuity can be estimated. Preferential looking does not always correspond with form visual acuity. It also may underestimate vision in infants with low vision.5 In infants with low vision, additional methods to measure visual function are recommended. Preferential looking is useful when visual acuity level can be quantified and in longitudinal management to detect change. In older children, visual acuity testing with Allen cards, the E-chart, or with numbers if the child does not know the alphabet can be used. In addition, parents are questioned as to the infant’s or child’s visual behavior. Questions should include whether the child exhibits an eye preference, a difference in visual behavior in light or dark environments, and whether there is ocular pain or avoidance of light. It may be difficult to determine visual acuity in each eye separately, particularly in the eye with poorer vision, because the infant may become uncooperative when the eye with better visual acuity is covered. In these cases, bilateral visual acuity or the vision in the better-seeing eye may be determined. The behavior of the child after having the better-seeing eye covered is noted in the record to indicate the suspicion of amblyopia. The infant’s visual system can be assessed objectively using electrophysiology. The visually evoked potential response (VER) assesses the optical pathways from the retina to the occipital cortex. The VER was reported as helpful in some diagnoses, such as albinism, where unilateral testing will show a largeramplitude response on the contralateral side than on the ipsilateral side in 70 to 100% of cases, reflecting the fact that the central 15 degrees of temporal retinal fibers decussate at the chiasm and travel to the contralateral lateral geniculate body instead of remaining ipsilateral.6,7 The pattern VER also can measure macular function and is associated with the level of visual acuity. FIGURE 20–2. Clinical pathway: Appearance of strabismus. OU, oculus uterque (both eyes). The electroretinogram (ERG) is used to evaluate retinal function. Any child with reduced vision, sensory nystagmus, or oculodigital stimulation (pressing on the eye) may be considered for ERG. The ERG amplitude continues to increase during the first year of life.8 In children and infants with cortical problems and delayed visual maturation, visual behavior can be reduced, even though the ERG and VER are normal. In these cases, it is possible for the vision to develop slowly. When a light is directed toward the eyes of a fixating child, the pupillary light reflexes should appear centered in each pupil. An eye that is turned out has exotropia, whereas an eye that turns in has esotropia. To detect which eye has a deviation, it is useful to have the child fix on an object while an occluder is brought first over one eye and then the other. As the occluder is removed from the eye, the examiner notes whether there is movement of the eye. The eye appears to move out when esotropia is present and when exotropia is present. An alternate cover examination will bring out latent strabismus. The occluder is swung from one eye to the other rapidly, and movement of the eyes is noted. The pupillary reflex can be used to estimate the amount of deviation (1 mm equals 7 degrees or approximately 15 prism diopters of deviation). Holding a prism bar in front of one eye as the alternate cover examination is performed provides a more accurate estimation. The power of the prism is adjusted during the alternate cover examination until there is no eye movement noted. In the infant and older child, it is possible to perform scleral depression without anesthesia. To perform a complete retinal examination of the posterior pole and peripheral retina with scleral depression, for example, in screening for retinopathy of prematurity (ROP), it is necessary to restrain a small infant, for example, swaddled in a blanket and have another person gently hold the head to minimize movement. A drop of anesthetic (proparacaine 0.5%) is placed into the conjunctival cul-de-sac and a lid speculum is inserted. The retina is then examined. For ROP, the posterior pole is examined first without applying additional pressure to the globe with scleral depression, and the retinal vessels are assessed for dilatation and tortuosity (Plus disease). Scleral depression can constrict retinal vessels and, when the pressure is removed from the globe, cause them to be dilated. The optic nerve is judged as to its color and size. The macular reflex is noted. Any vascular abnormalities or pigmentary irregularities are noted. The midperipheral and peripheral retina then are examined for the maturity of retinal vessel growth. In ROP, the stage and extent of the worst stage (clock hours) are determined. Scleral depression may be useful to examine the most peripheral retina. In eyes with zone 1 and some eyes with zone 2 ROP, scleral depression may not be necessary. In lightly pigmented eyes, the visibility of the underlying choroidal vessels may mislead the examiner into thinking that the retinal vessels have fully extended to the ora serrata when they have not. One way to reduce the chance of this error is to start at the optic nerve and follow the retinal vessels to the periphery. The retinal vascular arcades are often easier to see than peripheral retinal vessels. In the deeply pigmented fundus, the contrast between retina and its vasculature may appear poor, especially when using a 28- or 30-diopter lens. Examining these eyes using a 20–diopter lens may provide a better image in these cases. It is also useful to reduce external pressure on the globe so as not to constrict the retinal vessels and render them easier to see against a deeply pigmented fundus. An initial retinal evaluation of a child or infant should be planned to obtain the information necessary with minimal manipulation or trauma to the child. In complicated retinal evaluations, an examination under anesthesia (EUA) may still be necessary. The office examination is helpful in planning what is needed during the procedure under anesthesia. Once it is established that an EUA is necessary, a prolonged office examination of a fearful or uncomfortable child is usually unnecessary. The examination of the retina in a child takes patience, and it is helpful not to startle the child with sudden movements. It is important to speak to the child, especially one of an older age, and to be gentle. It may be helpful to use a pacifier to calm the infant during an eye examination. Infants and children may keep their eyes open more readily if the examiner avoids touching their lids or face with hands or fingers. The examiner can steady the arm, holding the condensing lens with the other hand while the child leans up against a parent. This makes it easier to focus light onto the child’s retina. It is useful to use low-light intensity of the indirect ophthalmoscope, particularly in the beginning of the examination, and it may be helpful to keep some light on in the examination room for the child who has fear of the dark. It is better to begin the retinal examination on the eye with poorer sight to get as complete an examination as possible before examining the fellow eye. If the vision in the fellow eye is normal, shining the light of the indirect ophthalmoscope into it may prevent the child from cooperating further with the examination of either eye. Sedation using chloral hydrate administered orally or rectally is used by some in children up to 3 years of age. The recommended dose of chloral hydrate is 50 mg per kilogram of body weight, up to a maximal single dose of 1000 mg.9 In small infants born prematurely, we do not recommend chloral hydrate sedation in the office because of the risk of apnea. We also avoid its use unless a parent or competent adult can carefully observe the child for the remainder of the day. In the older child, it is helpful to explain each step planned in the examination and not to mislead. When performing scleral depression, it is important not to press with the depressor perpendicular to the globe but rather to allow the movement of the globe to an extreme gaze to guide the depressor posteriorly and permit more tolerable depression of the far peripheral retina and ora serrata. It is also helpful to begin the examination of the peripheral retina in the superior and temporal quadrant and to end the examination at the superior and nasal quadrant. Depression in this quadrant may cause some discomfort, particularly if done before decompression of the globe has occurred from previous scleral depression. If a vitrectomy is planned, intraocular pressure is obtained with the Tono-Pen, avoiding the use of fluorescein and a handheld applanation tonometer so that visualization of the posterior segment is optimal for surgery. During an EUA, the anterior segments can be examined using the operating room microscope: The clarity of the corneas and the corneal diameters is recorded. The depth of the anterior chamber is noted. The presence of material such as fibrin or hemorrhage is noted in the anterior chamber. The iris is examined for its maturity and for abnormalities, such as transillumination defects, tumors, persistent tunica vasculosa lentis, or neovascularization. Cataracts, retrolental membranes, or subluxed lenses are noted. If the ciliary processes are pulled into the pupil, such as in persistent fetal vasculature (PFV) or persistent hyperplastic primary vitreous, this is recorded. The retina is examined with indirect ophthalmoscopy as described earlier in the office examination section. Increased magnification of retinal details can be achieved by using a flat contact lens placed on the cornea and the light from the operating room microscope. By increasing the magnification of the microscope, detail of the retina can be seen. The retinal field of interest can be brought into view by using a muscle hook to manipulate the globe. A 60-diopter lens also can be used to view the fundus. To focus on the retina, the microscope is moved anteriorly away from the cornea. Photographs of the retina can be taken with a camera mounted on the microscope using either of these methods (contact lens or 60-diopter lens) to view the fundus. If a vitrectomy is planned where a very clear cornea is needed, the contact lens is avoided. Fluorescein angiography can be performed in cases where suspected vascular leakage is expected. A contact wide-angle camera is also available.9a Ultrasonography can be performed to detect or characterize a retinal detachment, mass, or intraocular foreign body. This is performed in any infant or child with vitreous hemorrhage or where media opacity precludes a fundus view (Fig. 20–5), and can be done through the lid to avoid clouding the view through the cornea during surgery. Leukocoria is diagnosed by finding a whitish or dull rather than red reflex in the pupil when light is shined into the eye (Fig. 20–6). It may be secondary to media opacity at any level. Leukocoria or white pupil in the child should bring to mind several possible diagnoses (Fig. 20–3). These comprise mesodermal dysgenesis, including mutations of the PAX gene family,10 mucopolysaccharidoses (except II and III),11 mucolipidoses, fucosidosis, mannosidosis, Farber’s disease, congenital glaucoma, syphilis, rubella, cytomegalovirus, interstitial keratitis, fetal alcohol syndrome, birth trauma, dermoid,12 and persistent fetal vasculature, and others. These include lenticonus, congenital cataract from any cause including infectious (rubella, toxocariasis, toxoplasmosis, herpes simplex, cytomegalovirus, varicella zoster), inflammatory (juvenile rheumatoid arthritis, sarcoidosis), metabolic (hypocalcemia, galactosemia, galactokinase deficiency, diabetes mellitus, mannosidosis, phosphate, Lowe aminoaciduria, homocystinuria), traumatic (amniocentesis or other trauma), developmental (persistent fetal vasculature, aniridia), Alport syndrome, and others.13 This can occur with nonaccidental14 and accidental trauma, retinoblastoma,15 persistent hyaloid artery, persistent fetal vasculature, retinopathy of prematurity, protein C deficiency,16 Terson syndrome,17 disseminated intravascular coagulation disorder, or inflammation such as pars planitis or peripheral uveitis, and others.18 Inflammation can be caused by Toxocara canis infection,19 toxoplasmosis,20 peripheral uveitis,18 cytomegalovirus retinitis,21 endophthalmitis,22 tumor seeding, or hemorrhage from retinoblastoma, and others.23 This can be caused by Toxocara canis, tuberculoma, and sarcoid granuloma, and others. Choroidal coloboma is associated with renal abnormalities and has been described as an autosomal-dominant disorder caused by mutations in one copy of the PAX2 gene.24 These disorders include peripapillary staphyloma,25,26 optic-nerve coloboma, and morning-glory syndrome. Persistent fetal vasculature may be associated with cataract, vitreous hemorrhage, or retinal detachment, all of which may cause leukocoria. Norrie disease, incontinentia pigmenti, Coats’ disease, amniocentesis injury, uveitis, retinal detachment and X-linked retinoschisis may also be causes of leukocoria. Hypopyon, posterior or anterior synechiae, and cataract can be signs of inflammation or infection, whereas in disseminated retinoblastoma, the eye may be quiet, although seeded with tumor cells if glaucoma or neovascularization is not present. If the fundus is not visible, ultrasonography (Fig. 20–5) is performed to determine whether a tumor, retinal detachment, or vitreous debris is present. To detect retinoblastoma, calcification should be sought by ultrasonography or computed tomographic scanning; however, retinoblastoma, particularly those with a diffuse growth pattern, may not exhibit calcification.23,27 The finding of bilateral microcornea with cataracts may bring to mind congenital rubella, whereas unilateral microcornea with a retrolental membrane may be associated with persistent fetal vasculature. When persistent fetal vasculature occurs bilaterally, there may be an associated systemic condition. An ERG is helpful in the diagnosis of X-linked retinoschisis, with the finding of a reduced b/a ratio, and may be helpful in some mucopolysaccharidoses associated with retinal degeneration where an increased b implicit time is noted.11 A careful history that includes the family history is important in the workup. Tumors that occur in the pediatric age group may be malignant or benign (Table 20–1). Survival with retinoblastoma has increased with earlier detection and improved management. Retinoblastoma still presents challenges in diagnosis because it can present with a number of appearances, from classic leukocoria and calcified tumors to strabismus, vitreous hemorrhage, inflammation, iris neovascularization, hyphema, glaucoma, and endophthalmitis.15,23 It is useful to have a strong suspicion of retinoblastoma in unusual presentations in children. Although vitreous hemorrhage is not common in retinoblastoma, it is more unusual to occur with other retinal tumors. Shields et al recommended that vitreous hemorrhage with or without a retinal mass in a child or teenager should prompt the suspicion of retinoblastoma.15 Risk factors for metastasis include invasion into the orbit, choroid, or optic nerve.28,29 Surgical trauma from fine-needle biopsy and vitrectomy also can allow access of retinoblastoma into the orbit.15,30 It is important to obtain a second opinion, such as from an ocular oncologist, when in doubt. Classically, retinoblastoma is diagnosed on average at 13 months of age in bilateral cases and 24 months in unilateral cases. In the rare diffuse infiltrating retinoblastoma, presentation can occur later, after 5 years of age. The types of growth include exophytic, where the tumor grows into the subretinal space causing an overlying nonrhegmatogenous retinal detachment; endophytic, where the tumor grows into the vitreous cavity; and a diffuse infiltrating-type tumor that can mimic inflammation. Diagnosis is facilitated through ultrasonography and computed tomography (CT) scanning31 to detect calcification. The diffuse infiltrating form of retinoblastoma may lack calcification, however. In addition, astrocytic hamartoma, Coats disease,32 tuberculous, endophthalmitis,33 osseous fibroplasia associated with choroidal hemangioma, osteomas, and cyclitic membranes with bone formation may present with calcification.
Special Topics
20
Examination and Diagnosis of the Pediatric Patient
Pediatric Retinal Examination
What Are Signs of Retinal Disease in Infants and Children?
How Is the Visual System in Infants and Children Examined?
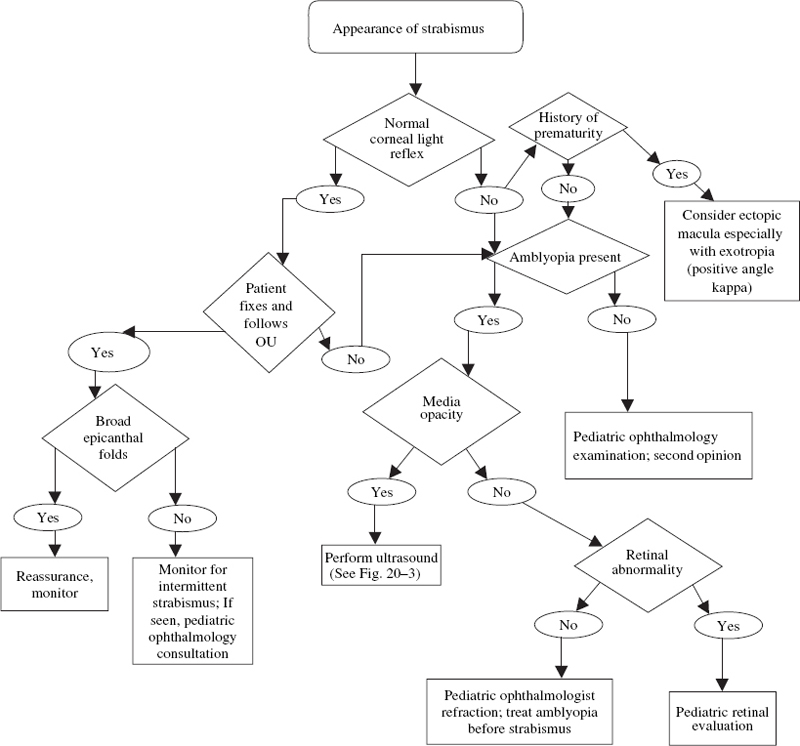
How Is Strabismus Detected?
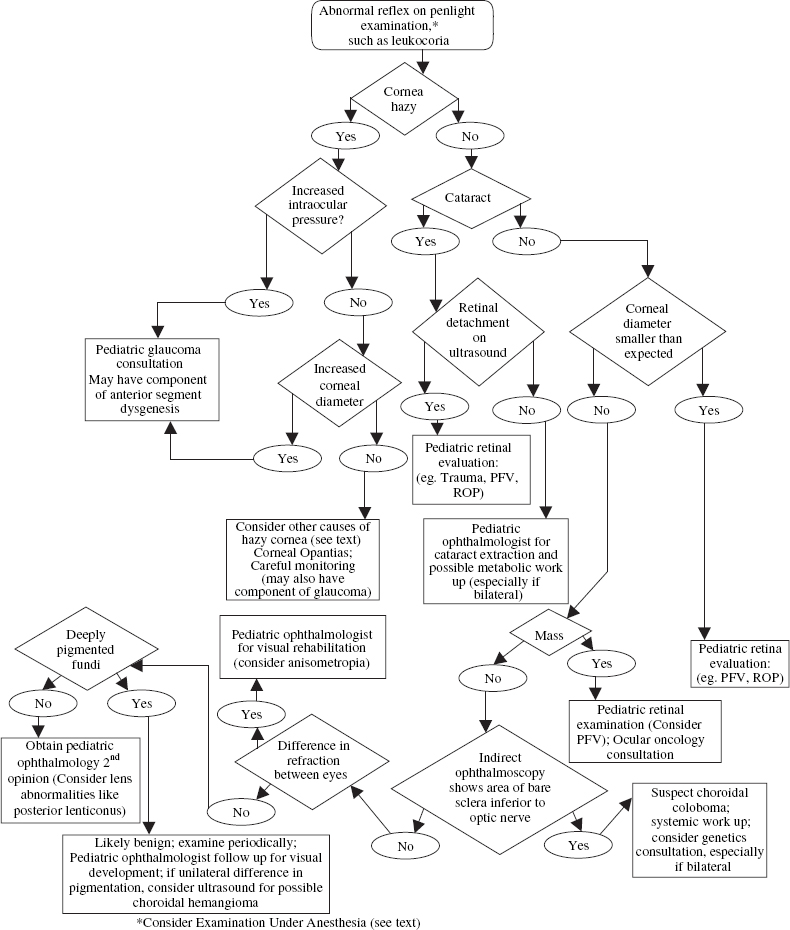
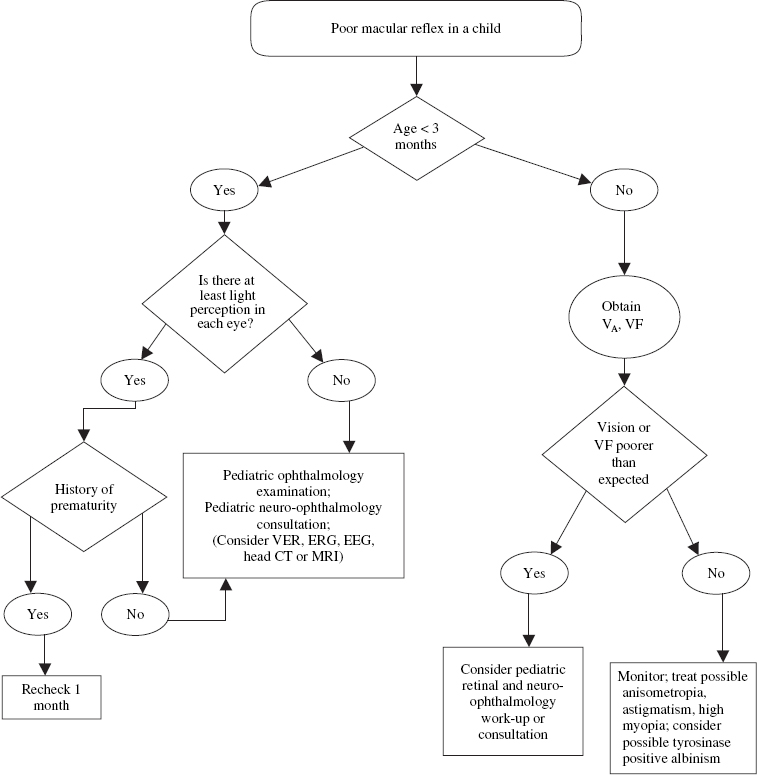
How Is the Retina in Infants and Children Examined?
What Is Done in the Examination under Anesthesia?
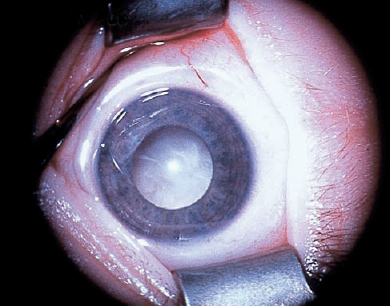
Leukocoria
What Is the Differential Diagnosis of Leukocoria?
CORNEAL OPACITIES
LENS ABNORMALITIES
VITREOUS HEMORRHAGE
INFLAMMATION
GRANULOMA
CHOROIDAL COLOBOMA
OPTIC-NERVE DISORDERS
VITREORETINAL DISORDERS
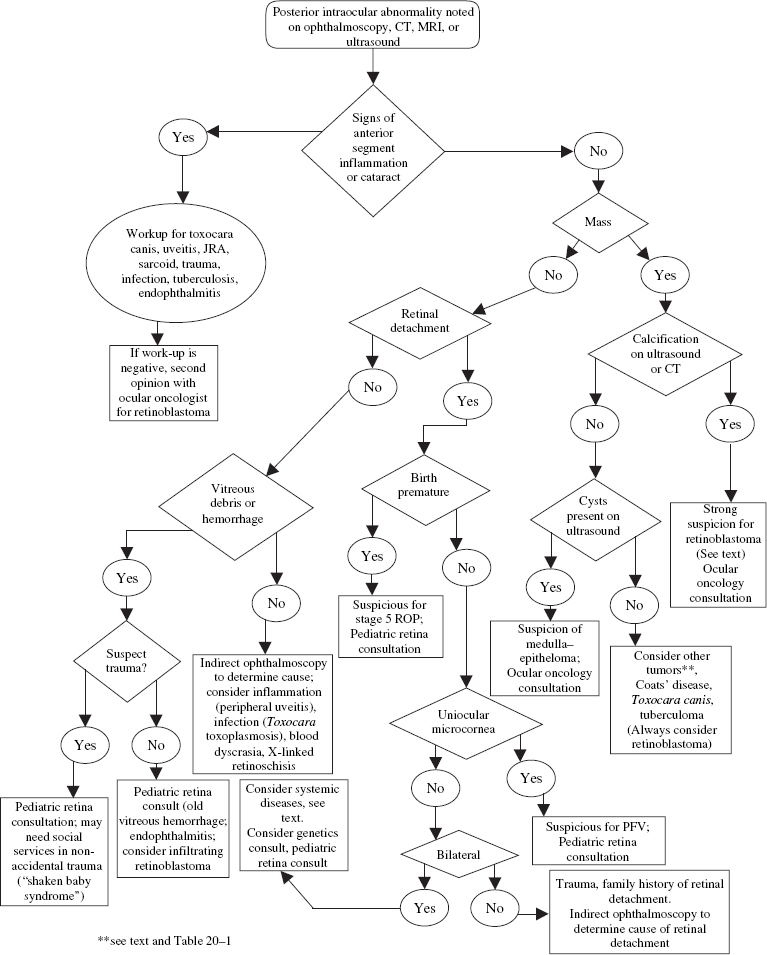
What Are Some Tumors of the Posterior Segment in Children?
Retinoblastoma
| Malignant tumors of the retina | Retinoblastoma |
| Medulloepithelioma (dictyoma) | |
| Adenocarcinoma (rare in children) | |
| Malignant tumors of the uvea | Melanoma (extremely rare in children) |
| Benign uveal tumors | Choroidal hemangioma |
| Choroidal osteoma | |
| Neurofibroma, neurilemmoma, leiomyoma (often in neurofibromatosis type 1) | |
| Benign retinal tumors | Astrocytic hamartoma |
| Retinal hemangioblastoma | |
| Congenital hypertrophy of the retinal pigment epithelium | |
| Combined hamartoma of the retina and retinal pigment epithelium | |
| Optic nerve glioma | Often in neurofibromatosis type 1 |
Data from Damato B. Ocular Tumors: Diagnosis and Treatment. Oxford, England: Butterworth-Heinemann; 2000, with permission.
Retinoblastoma occurs because of a mutation in the RB gene located on the long arm of chromosome 13 at region 14. The gene normally suppresses cell division. Malignancy occurs only when both the paternal and maternal genes are malfunctioning. A somatic mutation (60%) occurs in a single cell and can lead to a solitary retinoblastoma, whereas a germline mutation (40%) produces a mutation in every cell in the body. Germinal mutations cause bilateral retinoblastomas in 75% of patients, unilateral retinoblastoma in 15%, and no tumors in 10%. Germline mutations also can cause a rare midline intracranial primitive neuroectodermal tumor (PNET). These usually occur 2 to 4 years after the diagnosis of bilateral retinoblastoma. The prognosis is poor in PNET, although chemotherapy and radiation treatment of small asymptomatic tumors have improved survival.34 Detection remains a problem. In one series of 226 patients, neuroimaging with CT scans of the orbits and magnetic resonance imaging (MRI) of the head did not improve detection of PNET.35
Treatment of retinoblastoma includes chemotherapy, photocoagulation, transpupillary thermotherapy, cryotherapy, brachytherapy, external beam radiation, and enucleation.36
Secondary tumors are usually from osteosarcoma of the skull and long bones, cutaneous melanoma, and sarcomas. Secondary tumors may be more common after chemotherapy or radiation treatment.
Medulloepithelioma
Medulloepithelioma is a rare sporadic embryonic tumor, usually arising from the ciliary body and most often unilateral.37,38 It is derived from embryonic nonpigmented ciliary epithelium and may be histopathologically indistinguishable from retinoblastoma. Cystic spaces filled with hyaluronic acid and undifferentiated neuroblastic cells may provide aid in the diagnosis.39 Death occurs from intracranial invasion or rarely from metastatic disease.37 The anterior presentation of medulloepithelioma in the eye differentiates it some from retinoblastoma. Distinguishing ultrasonographic characteristics include cystic changes, mild internal vascularity, and absence of calcification.40 Treatment may include surgical excision or enucleation.
Choroidal Hemangioma
Choroidal hemangioma41 in children is rare but when present is often the diffuse type that is associated with Sturge–Weber syndrome. The hemangioma tends to cause a bright red pupillary reflex. Within the fundus, the choroid appears thickened and is often red-orange in appearance (Fig. 20–7). The diagnosis may be aided by examining the fellow eye, which will appear duller in contrast. Pigment stippling and dilated tortuous retinal vessels cover the lesion. Vision is affected because the hemangioma leaks causing exudative retinal detachments. Secondary glaucoma and cataract can occur. Sturge–Weber syndrome also can lead to increased episcleral venous pressure and glaucoma. Other findings with Sturge–Weber syndrome include nevus flammeus, iris heterochromia, conjunctival and episcleral telangiectasia, ocular melanosis and leptomeningeal disease leading to seizures. Fluorescein angiography reveals early hyperfluorescence and late leakage of the hemangioma. Ultrasonography reveals high internal reflectivity without acoustic hollowness. Treatment includes observation for asymptomatic tumors and external-beam radiotherapy, brachytherapy, proton-beam radiotherapy, phototherapy, and possibly photodynamic therapy when vision is threatened or reduced.
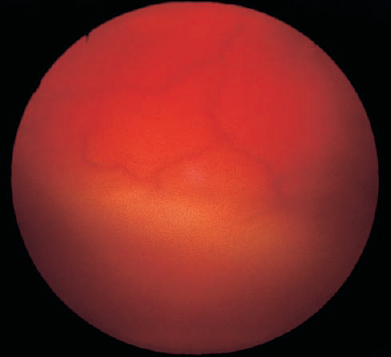
FIGURE 20–7. Diffuse choroidal hemangioma and serous retinal detachment in patient with Sturge–Weber syndrome. Note red appearance of hemangioma in the superior aspect of image and opaque retinal fold in the inferior aspect of image.
Choroidal Osteoma
Choroidal osteoma can be detected in children, although it occurs more commonly in early adulthood.42 It may appear white, yellow, or orange and often is located adjacent to the disc or macula. Osteomas are bilateral in 25% of cases. They may be asymptomatic unless the fovea is involved by the tumor or if choroidal neovascularization occurs. Fluorescein angiography reveals diffuse irregular hyperfluorescence. Ultrasonography reveals a highly reflective anterior surface with a shadow cast into the orbit.
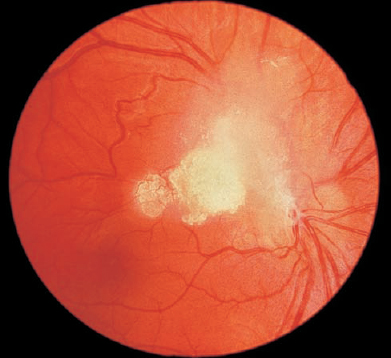
FIGURE 20–8. Solitary astrocytic hamartoma in the right eye of a young boy without tuberous sclerosis. Visual acuity was 20/25. (Photograph taken by P. Buch, CRA)
Astrocytic Hamartoma
These tumors can be solitary (Fig. 20–8) or multiple. Multiple lesions occur with tuberous sclerosis, an autosomal-dominant disease caused by mutations located at 9q34 and 16p13.43,44 Ocular abnormalities occur in 50% of patients with tuberous sclerosis and include angiofibromas of the eyelid skin and conjunctiva, colobomas of iris and lens, megalocornea, and white lashes.
Astrocytic hamartomas may also occur in neurofibromatosis-1. Most astrocytic hamartomas do not require treatment. Complications from vitreous hemorrhage, neovascular glaucoma, and vitreous seeding rarely occur.45
Retinal Hemangioblastoma
These occur in approximately two of three patients with autosomal-dominant von Hippel–Lindau disease.46 Symptoms usually occur in teenage years and twenties, but lesions can be detected in childhood.
The small angiomas leak and can grow, leading to exudative retinal detachment, traction retinal detachment, and blindness. These tumors are best treated when they are small, and so early detection with careful examination using a Goldmann three-mirror lens is important. Treatment of small retinal angiomas is performed with photocoagulation and cryotherapy.47 Larger lesions may require brachytherapy or vitrectomy and endolaser. Life-long screening for associated cerebellar hemangioblastomas (25% of patients with retinal lesions), renal cell carcinoma, spinal canal hemangioblastoma, pheochromocytoma, islet cell carcinoma of the pancreas, polycythemia, and visceral cysts is important.
Congenital Hypertrophy of the Retinal Pigment Epithelium
When multiple, these asymptomatic lesions have been termed bear tracks. Solitary lesions are usually benign, but multiple bear tracks can be associated with familial adenomatous polyposis (mutation in chromosome 5), in which malignant transformation of polyps have been reported later in life.48
Combined Hamartoma of the Retina and Retinal Pigment Epithelium
These are usually detected in young adults but can be detected in children, sometimes causing sensory strabismus.49 The hamartoma of glial cells, blood vessels, and retinal pigment epithelial cells invades the retina (Fig. 20–9) and may cause choroidal neovascularization, vitreous hemorrhage, epiretinal membrane, retinal contracture, wrinkling, and macular edema.
Arteriovenous Malformation
Arteriovenous (AV) malformations are nonhereditary, unilateral, and usually asymptomatic. They have been associated with macular preretinal membranes, vitreous hemorrhage, and central and peripheral venous occlusions leading to neovascular glaucoma.50
Coats’ Disease
This unilateral sporadic disease usually affects males and has associated dilation of retinal vessels with exudation. Early on, exudates are deposited in the retina, where subretinal fluid is reabsorbed, often distant from the telangiectatic leaking vessels. The deposition may occur in the posterior pole (Fig. 20–10). When macular exudates are noted, the peripheral retina should be examined for vascular anomalies. Treatment of the leaky vessels with photocoagulation or cryotherapy can reduce leakage of telangiectatic vessels, and over time, fluid and exudates are reabsorbed.47 Treatment may need to be repeated, especially when closing leaking arterioles. Visual acuity often remains poor once macular exudation occurs. Early treatment before the development of macular exudates is recommended, but detection may be difficult. There is no familial tendency, and the first sign in infants and young children may be sensory strabismus.
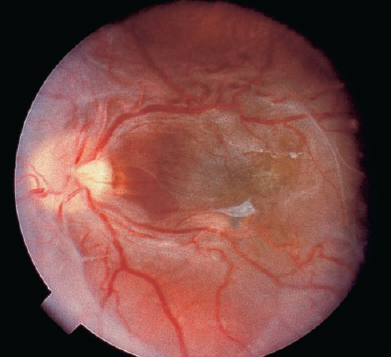
FIGURE 20–9. Combined hamartoma of the retina and retinal pigment epithelium.
Late Coats’ disease can be associated with exudative retinal detachments, preretinal membranes, and subretinal organization. Coats’ disease can be mistaken for exophytic retinoblastoma in that both can be associated with a retinal detachment, subretinal mass, and abnormal retinal vessels. The average age at diagnosis of Coats’ disease is 3 years, whereas for unilateral retinoblastoma it is 18 to 24 months. Even at age 1 year, however, exudative Coats’ disease has been diagnosed, and older children have presented with retinoblastoma.32 In addition, calcification, a hallmark in retinoblastoma, can occur in Coats’ disease and is often absent in diffuse infiltrating retinoblastoma.
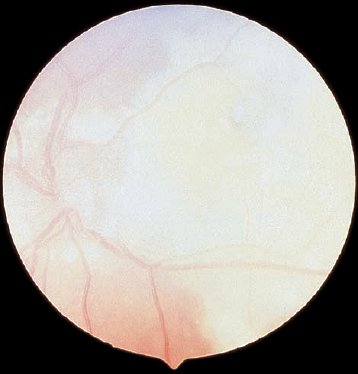
FIGURE 20–10. Coats disease in a male child. (Photographtaken with Kowa camera by M. Maio, CRA)
Patients with Coats’ disease should receive timely treatment and should be followed up periodically and treated for new areas of leakage.
What Are Some Causes of Differences in Ocular Size in Infants and Children?
An enlarged cornea may be associated with congenital glaucoma in a child under 3 years of age. Glaucoma may be secondary as well, such as that associated with retinoblastoma and uveitis. Mesodermal dysgenesis also may be associated with an enlarged cornea, staphyloma, and glaucoma. Buphthalmos51 and congenital megalophthalmos syndrome have been associated with retinal detachment and giant retinal tears.
A small corneal diameter is associated microphthalmos, with persistent fetal vasculature, retinopathy of prematurity, coloboma, and other genetic syndromes.
What Are Some Causes of Retinal Detachment in Infants and Children?
Genetic Causes
NORRIE’S DISEASE
Norrie disease causes bilateral blindness and has an X-linked recessive transmission.52,53 Therefore, it affects males only, the mother being the obligatory carrier. Blindness is due to retinal detachment, which usually occurs within a few months after birth. Mental retardation may occur in 25% or more of patients and about a third develop progressive sensorineural deafness.54 Patients often present with bilateral retinal detachment, exudative or traction, and blindness.
In early disease, there is often a persistent hyaloid artery and an absence or minimal development of retinal vessels. Evidence from retinal histopathology in genetically altered mice supports the line of thinking that Norrie’s gene may be necessary for the regulation of retinal vascular development.55,56
INCONTINENTIA PIGMENTI
Incontinentia pigmenti is a rare disease that is transmitted through the mother’s mitochondria. It appears X-linked dominant. Only females with one gene survive. Early, the retinal vasculature is not fully developed in the peripheral retina, leading to large avascular peripheral zones, similar to that seen in retinopathy of prematurity. Later, extraretinal neovascularization at the interface of the avascular and vascularized retina proliferates and leads to traction retinal detachments.57 Incontinentia pigmenti is often bilateral, but it may be asymmetric. It is important to be aware of the early stages of incontinentia pigmenti because laser to the peripheral avascular retina, when extraretinal neovascularization is present, may prevent further progression of the disease. Dermatologists should notify the ophthalmologist of an infant with skin lesions because these precede the retinal detachment. The skin findings appear soon after birth and appear as a linear eruption of bullae that predominantly affects the extremities. The bullae gradually resolve to leave a linear pattern of pigmentation.
FAMILIAL EXUDATIVE VITREORETINOPATHY
Familial exudative vitreoretinopathy was first described by Criswick and Schepens.47,58 The condition is often bilateral, autosomal dominant, and is thought to involve the inner retina, particularly its maturation to the ora serrata. The condition is similar in appearance to ROP, but patients are not premature or of low birth weight. Clinically, there is a temporal avascular zone, often with leakage of the vessels between the vascularized and avascular retina. Later, serous retinal detachments with exudation can occur. Traction retinal detachment can also occur. Vitreous surgery for retinal detachment may be beneficial.59
STICKLER SYNDROME (WAGNER–STICKLER SYNDROME)
Stickler syndrome is a dominantly inherited disorder of collagen connective tissue with ophthalmic, orofacial, auditory, and articular manifestations. Orofacial characteristics include a flat midface with a depressed nasal bridge, reduced nasal protrusion, anteverted nares, and micrognathia. About 25% of patients may have some midline clefting. Deafness may be sensori-neural or conductive from serous otitis media. Joint hypermobility and later degenerative arthropathy occurs.
Stickler syndrome is the most common inherited cause of rhegmatogenous retinal detachment in childhood. Most eyes, but not all, have early onset of high myopia. Congenital cataracts, vitreous abnormalities and veils, and perivascular pigmented lattice degeneration are associated. Anomalies in the anterior chamber angles can predispose to glaucoma. Several gene defects in type II collagen60 and type XI collagen61 have been reported in Stickler phenotypes.
The retinal detachments in Stickler syndrome are often associated with giant retinal tears and large tears along areas of lattice degeneration.62 The lattice degeneration in Stickler syndrome can be perivascular and, therefore, extend radially from equatorial to posterior retinal locations. Retinal reattachment with scleral buckling may be difficult because of this, and vitrectomy is often necessary.
MARFAN SYNDROME
This autosomal-dominant syndrome is associated with abnormalities in elastin and collagen with cardiovascular abnormalities, such as dilation of the aortic root with dissecting aortic aneurysm, heart valve abnormalities, aortic regurgitation and mitral valve prolapse. Patients tend to have long, slender arms and legs, long fingers, and hypermobility of the joints, a longer lower torso than normal, and pectus excavatum. Ocular findings include enlargement of the globe,63 ectopia lentis, filtration angle abnormalities and glaucoma, thin sclera, poorly dilating pupils, and pigmentary changes in the retinal periphery. Early vitreous syneresis and retinal breaks and detachment occur. Bilateral retinal breaks and detachments are reported in more than 50% of cases.64 Removal of subluxed lenses via vitrectomy and lensectomy is successful, and retinal detachment is reduced in cases with intraoperative prophylactic laser treatment of lattice degeneration.65
EHLERS–DANLOS SYNDROME
Ehlers–Danlos syndrome consists of a group of hereditary connective tissue disorders caused by defective collagen synthesis, the main features being hyperelasticity and vulnerability of the skin, recurrent bleeding from fragile blood vessels, and secondary deformities of the joints. Ocular involvement is a rare occurrence,63 for example, corneal and scleral rupture from minor blunt injury, lens displacement, and rhegmatogenous retinal detachment. Treatments with conventional scleral buckling surgery have been reported, in some instances using buckle loops glued onto thin sclera with cyanoacrylate.66 Vitrectomy was reported with the complications of pronounced choroidal detachment and bleeding, difficulty closing the sclerotomies as a result of scleral thinning, and later anterior proliferative vitreoretinopathy with reduced vision. Surgical procedures other than primary vitrectomy should be considered.67
ANIRIDIA
Aniridia may present sporadically or as an autosomal-dominant condition. Typical ocular findings are lens subluxation, cataract, hypoplasia of the fovea and optic nerve, and glaucoma. Also reported is an association with retinal detachment and giant retinal tears in eyes that become buphthalmic.68
Developmental Causes
CHOROIDAL COLOBOMA
During the fifth to seventh week of normal development, the fetal or choroidal fissure closes by fusion of each side of the optic cup.69 This process begins at the equator and proceeds toward the anterior aspect of the eye and toward the optic nerve. Failure of the closure of the fetal fissure can produce a variety of colobomatous lesions affecting the optic nerve, retina, choroid, lens, and iris. The location of the fetal fissure is inferior and nasal to the optic nerve. Thus, the location of developmental colobomas tends to be inferior and nasal.
A choroidal coloboma may produce a white pupillary reflex. Dilated funduscopy is performed to rule out a retinal detachment. Retinal detachment can occur secondary to a break outside of and unrelated to the coloboma70 or from vitreoretinal traction and retinal breaks within the coloboma (Fig. 20–11). In the first case, standard scleral buckling leads to repair of the retinal detachment. In the second case, more complicated vitreoretinal surgery is required.71,72
A combination of four of the following features has been termed CHARGE syndrome73: coloboma of the eye, heart defects, atresia choanae, retardation of growth or development, genital anomalies, and ear anomalies with or without deafness. The presentation of the features may vary in severity.
LENS COLOBOMA
In infants and children, retinal detachment may be associated with a lens coloboma, often inferior and nasal in location, and associated with absence of zonular fibers in the area of the coloboma. Tissue may connect the posterior lens capsule with the nasal retinal periphery of the retina, causing persistence of an embryonic fold of redundant retina in the extreme fundus periphery. Such an adhesion may cause a giant retinal tear that starts nasally and may extend. When the peripheral retina is adherent to the lens 360 degrees, nonattachment of the retina results. Lens coloboma is generally bilateral.71,74
PERIPAPILLARY STAPHYLOMA
Peripapillary staphyloma25 is nonhereditary, usually unilateral, and usually associated with markedly reduced visual acuity. Cases exist with good visual acuity if the macula has not been distorted. A retinal detachment may occur within the staphyloma. Detection of the retinal detachment may require ultrasonography.26 The macular reflex is often noted to be closer to the optic nerve than usual, or it may be hidden within the staphyloma.