Chapter 28 Craniofacial abnormalities
Introduction
This chapter discusses two main groups of disorders:
1. Craniosynostoses, in which premature closure of sutures causes an abnormally shaped skull, e.g. Crouzon’s and Apert’s syndromes.
2. The clefting syndromes, in which there is a failure of apposition or fusion of fetal tissues. These include the mandibulofacial dysostoses, e.g. Treacher-Collins and Goldenhar’s syndromes.
Craniosynostosis
Craniosynostosis is premature fusion of one or more cranial vault sutures with resultant skull deformity. It occurs in approximately 1 in 2500 births and may be primary or secondary. Thirty percent of cases are syndromic, usually with multiple suture involvement and associated primary malformations in the face, trunk, or extremities. Syndromic craniosynostosis patients present with significant cosmetic challenges and face complex neurologic, ophthalmologic, and airway difficulties. Non-syndromic (“simple”) craniosynostosis may have neurologic or ophthalmologic complications. Some cases of simple craniosynostosis represent the mild end of a spectrum of syndromic disease.1
Pathophysiology
Pathogenesis and genetics1
1. Tyrosine-kinase receptors: FGFR1, FGFR2, and FGFR3
3. TWIST1 and MSX2, ligand receptors
4. EFNB1 and EFNB2 and intracellular cell membrane protein trafficking: RAB23.
Five of these mutations are involved in cell proliferation and ossification and belong to a common molecular pathway. Gain-of-function mutations represent the main molecular mechanism causing the disorders, but loss-of-function may lead to the phenotype. These seven genes account for 30% of syndromic cases. The genetic etiology of non-syndromic craniosynostosis is poorly understood; familial occurrence and associated gene changes within fused sutures are the same for both syndromic and non-syndromic cases.2 The reader is referred to genetic databases such as the Online Mendelian Inheritance in Man (OMIM) Database (http://www.ncbi.nlm.nih.gov/entrez). Environmental factors have also been implicated in the etiology of craniosynostosis, including maternal smoking and altitude and intrauterine head constraint.3 Craniosynostosis may occur secondary to various conditions, including shunted hydrocephalus, metabolic disorders such as hyperthyroidism and rickets, and hematologic disorders such as thalassemia and sickle cell anemia.3
Effects on the skull
Cranial sutures are fibrous joints providing a malleable quality to the head, allowing vaginal birth and growth of the brain during early development. Premature skull suture fusion results in restricted cranial growth perpendicular to the fused suture and compensatory growth at the remaining open sutures (Fig. 28.1). The principal clinical manifestations have been given Greek or Latin descriptors; identification of the involved suture has found more favor recently. Trigonocephaly (“triangular head”) is metopic suture synostosis, scaphocephaly (“boat shaped head”) is sagittal suture synostosis, plagiocephaly (“twisted head”) is either unilateral coronal or unilateral lambdoid suture synostosis, and brachycephaly (“short head”) is bilateral coronal suture synostosis (Table 28.1). Multiple suture involvement results in more complex head morphologies such as tryphyllocephaly (Kleeblatschadel, clover leaf, or trilobed skull) or oxycephaly (“towering head”). Premature synostosis also occurs in the cranial base and facial skeleton. Cranial base underdevelopment compounds primary mid-facial hypoplasia through its indirect effects on mid-facial growth. The resulting abnormalities of palate, dentition, airway, and hearing may be severe.
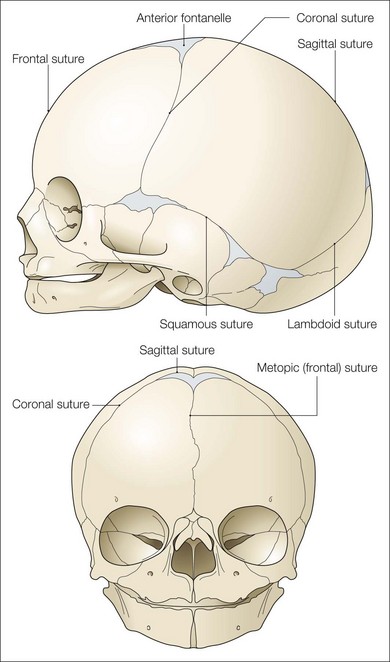
Table 28.1 Simple craniosynostosis
| Suture involved | Features |
|---|---|
Metopic 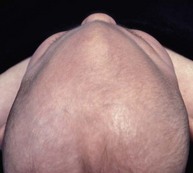 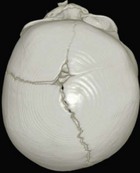 | |
Unilateral coronal  | |
Sagittal 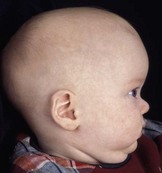 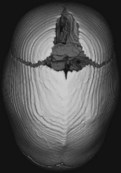 | |
Unilateral lambdoid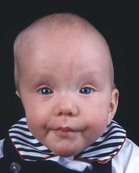 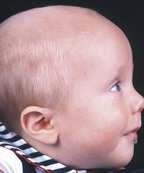  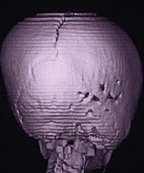 |
Effects on intracranial pressure, the brain, and optic nerve
Brain development is intimately tied to growth of the skull; children with craniosynostosis may have developmental delay.4,5 Reported incidences vary widely. Increased intracranial pressure (ICP) may occur in 75% of syndromic patients and 20% of single suture craniosynostosis patients. In syndromic patients four dynamic factors may contribute to raised ICP: restricted skull volume (“craniostenosis”), intracranial venous hypertension, hydrocephalus, and sleep apnea caused by facial deformity.6 Due to its slowly progressive nature, the majority of patients have no warning symptoms. Radiologic findings are not necessarily related to intracranial pressure recordings and optic nerve swelling is frequently absent. FGFR gene products are found in the optic nerve sheath and, when these are mutated, its failure to dilate may be due to abnormal fibrous tissue in the optic nerve sheath or lamina cribrosa.7 This increased resistance and the limited autoregulatory capacity within the sheath increases the susceptibility of the optic nerve to decreased cerebral perfusion pressure due to raised ICP, venous hypertension, and obstructive sleep apnea-related CO2 retention.8 Sleepapnea-related hypoxia may also contribute to brain and optic nerve damage.
Effects on the orbit
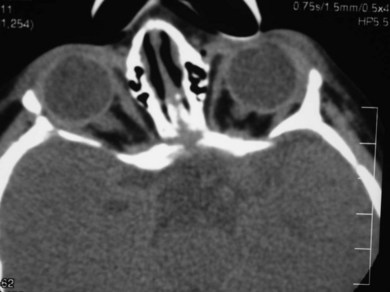
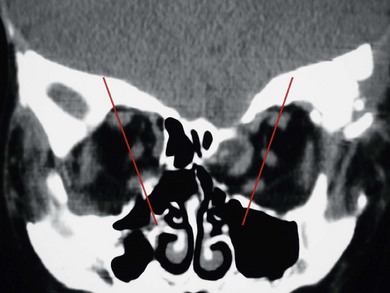
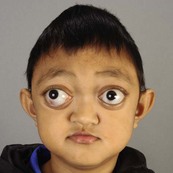
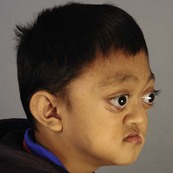
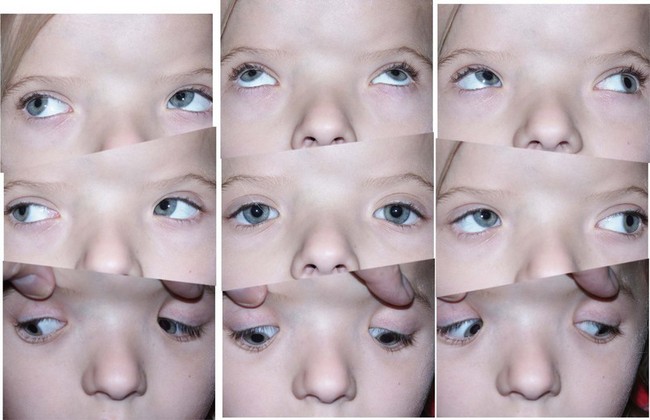
In syndromic craniosynostosis mid-facial hypoplasia and underdevelopment of the skull base lead to shallowing of the orbits, retrusion of the orbital rims, and proptosis (Fig. 28.2). Vision threatening corneal exposure or globe prolapse may occur. Keratinocyte growth factor receptor is a splice variant of FGFR2 and corneal epithelial healing may be abnormal with some FGFR mutations. Lowering of the cribriform plate and anterior cranial fossa floor with overgrowth of the ethmoid complex results in failure of anterior rotation of the orbital axes in fetal life (see Chapter 2), creating divergent orbital axes and hypertelorism (see Chapter 22). This divergence may be greater in the frontal than maxillary planes, resulting in ex-cyclorotation of the orbits (Fig. 28.3). The external manifestation of this is the anti-mongoloid palpebral fissure. Premature closure of the metopic suture may result in hypotelorism. These changes in orbital axis and angulation increase the likelihood of strabismus, which is seen in up to 90% of patients. FGFR2 is expressed in the extraocular muscles (EOMs); this may explain some of the EOM abnormalities, including bifid, hypoplastic or absent muscles, microscopic abnormalities, and a gristly, inelastic feel.7 FGFR2 is also expressed in the embryonic rat lens and cornea. This may account for the high incidence of refractive error in these patients.
Management
Diagnosis2
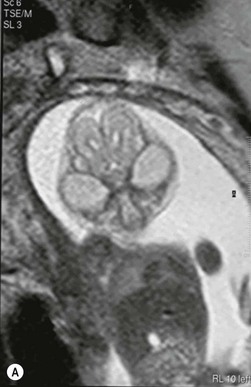
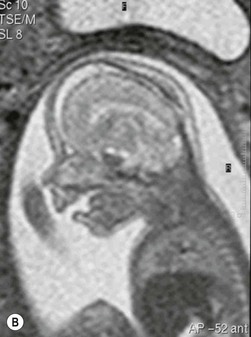
Simple craniosynostoses are summarized in Table 28.1 and the most common craniosynostosis syndromes are described in Table 28.2. Syndromic craniosynostosis presents little challenge to detection and may be diagnosed in utero (Fig. 28.4). There are a number of differential diagnoses to consider with simple synostoses. History and examination will usually resolve any uncertainty. Attention should be paid to:
Table 28.2 The most common craniosynostosis syndromes
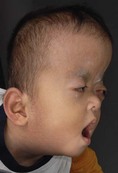
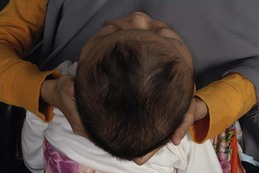
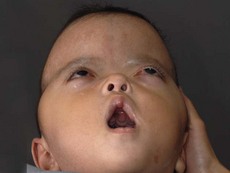
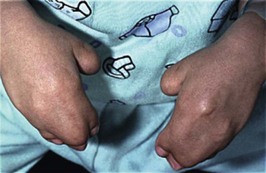
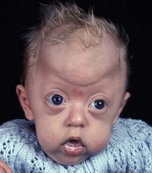
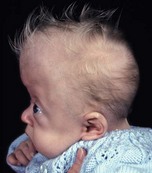


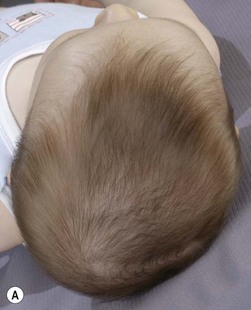
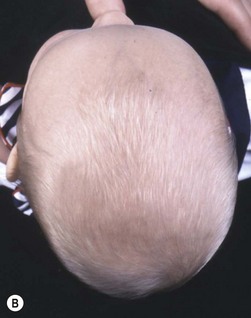
The sutures may be palpated for abnormal ridging or immobility. Deformational plagiocephaly, a common condition since the introduction of the “back to sleep” intervention to reduce the risk of sudden infant death syndrome, may be confused with lambdoid synostosis, which is very rare. Deformational plagiocephaly is characterized by a history of development of the head shape in the first few months of life in association with persistent positioning on one side, torticollis, or an inactive or developmentally delayed infant. The resultant occipitoparietal flattening may be differentiated from unilateral lambdoid synostosis by a parallelogram shaped head seen from above and anterior displacement of the ipsilateral ear. Unilateral lambdoid synostosis results in a trapezoidal shaped head from above and the ear is posteriorly displaced (Figs 28.5 and 28.6). The relatively large head and poor neck muscle tone of the premature baby results in a laterally turned head and may result in a long narrow head. The distinction from sagittal synostosis can be made by the mobile sagittal suture and correction of the head shape at 3 months of age as head control increases. Metopic synostosis may present some diagnostic difficulty; it is a spectrum of disease: at one end is the development of a metopic ridge in the first year of life, which usually requires no treatment. This may be differentiated from premature closure of the metopic suture seen in primary microcephaly by normal head circumference. At the other end of the spectrum is classic trigonocephaly characterized by metopic ridging in association with supraorbital recession and hypotelorism (see Chapter 22).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


