Chapter 33 Corneal abnormalities in childhood
The white cornea at birth poses an important differential diagnosis. The first consideration is congenital glaucoma (see Chapter 37). Intraocular pressure is elevated and the corneal diameter is large. Ruptures in Descemet’s membrane may be present. The optic nerves show increasing cupping, often reversible with intraocular pressure control. Urgent intervention is usually indicated.
Forceps injury is another cause of corneal clouding apparent at birth. Forceps marks may be visible on the lids or cheek. A linear, usually vertical, rupture of Descemet’s membrane is present causing corneal edema which always resolves leaving astigmatism. Late corneal decompensation is possible. Metabolic disorders that may cause congenital corneal clouding such as the mucopolysaccharidoses are discussed in Chapter 62; congenital hereditary endothelial and stromal dystrophies are discussed in Chapter 34.
Developmental defects
Embryologic errors
Anterior segment dysgenesis (Chapter 32)
Anterior segment dysgenesis is a spectrum of congenital anterior segment disorders that result from abnormal migration of the waves of neural crest that, from the sixth week of gestation, invade the primary mesenchyme behind the surface ectoderm, giving rise to the corneal endothelium, corneal stroma, angle, and the iris stroma.1 Thus, anterior segment dysgenesis frequently leads to glaucoma and corneal opacification.
The anterior segment dysgeneses2–8 have been linked to abnormalities in homeotic genes that control migration and differentiation of neural crest; these include mutations in the PAX6, PITX2, and FOXC1 genes.2,3,4
Penetrating keratoplasty for unilateral cases is of limited benefit since the risk of graft failure and amblyopia is high, but may be considered more readily in bilateral cases. Sometimes, penetrating keratoplasty is performed on the eye with more severe opacification, while observing the less affected eye for spontaneous clearing. The success of penetrating keratoplasty in this age group and for this condition is limited; satisfactory visual results are seen in less than 50% of cases due to graft failure, amblyopia, or glaucoma.9
Genetic syndromes
Trisomy 18 and trisomy 8 mosaic
Dense corneal opacity has been reported in trisomy 8 mosaic, described as richly vascularized fibrous tissue localized to the superficial layers of the cornea.10 Spontaneous resolution of this opacity has been observed, but other ophthalmic hallmarks of the disorder including diffuse retinal pigment abnormalities, attenuated ERG, Duane’s syndrome, and macular hypoplasia persist (Fig. 33.1).
Ectodermal dysplasia
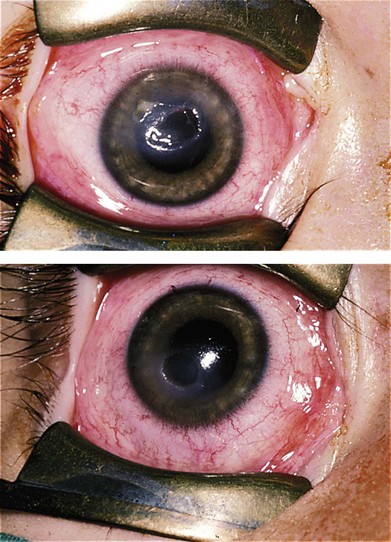
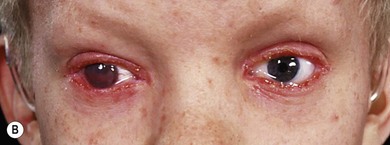
Ectodermal dysplasia is a rare (1 : 100 000 live births), usually X-linked or autosomal recessive, condition with abnormal eccrine glands, wispy or absent hair, and abnormal teeth or nails. Numerous syndromes make up the ectodermal dysplasia group; the two main groups are the hidrotic and the anhidrotic (or hypohidrotic) forms. Ocular involvement is usually limited to anhidrotic forms.11
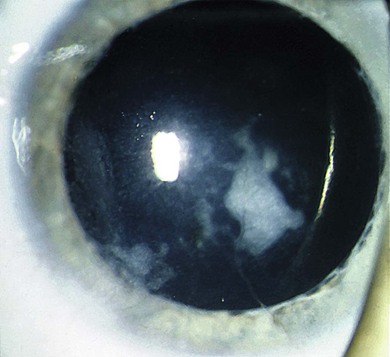
Ocular abnormalities include blepharitis, conjunctivitis, corneal scarring, pannus, photophobia, decreased tear production, and entropion with trichiasis12,13 (Fig. 33.2). Meibomian and lacrimal abnormalities may lead to ocular surface disease resulting in keratopathy, infection, and scarring14 (Fig. 33.3). However, recurrent erosions, pannus, and opacification occur with normal tear film measures; this suggests that the primary pathology may sometimes be limbal stem cell insufficiency. Thus, conjunctival limbal allograft has been suggested for corneal opacification associated with pannus.15
Corneal opacities associated with dermatologic conditions
Ichthyosis
The ichthyosiform dermatoses are a group of disorders characterized by scaling. “Harlequin baby” and “collodion baby” are extreme congenital forms that may have congenital ectropion.16 They frequently succumb to skin infections in the neonatal period. Ichthyosis vulgaris is the most common form, inherited as an autosomal dominant trait, with scaling of the extensor surfaces and back. No eye problems occur.
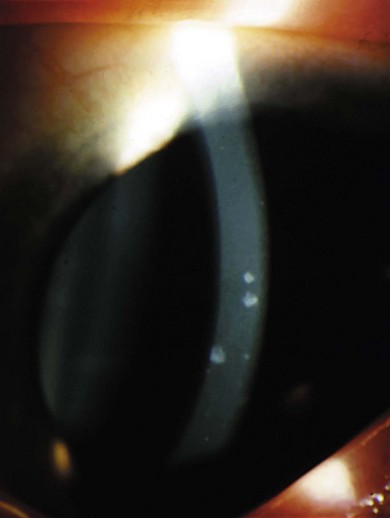
X-linked ichthyosis is congenital and occurs in 1 in 6000 males.17 Afflicted individuals suffer scaling of the scalp, face and neck, abdomen, and limbs; palms and soles are spared. Corneal nerves may be thickened and band keratopathy occurs as an isolated abnormality.18 Superficial corneal lesions, which stain with fluorescein (Fig. 33.4A), occur; they are usually transient but recur and eventually cause superficial scarring. The scarring and superficial lesions may be caused by eyelid abnormalities, or may occur independently.
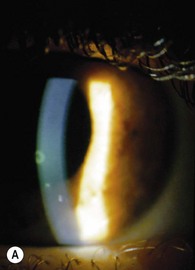
Small posterior corneal opacities are located in deep corneal stroma or Descemet’s membrane and are not visually significant.18
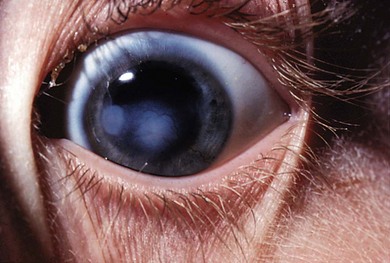
Lamellar ichthyosis and ichthyosis linearis circumflexa are severe autosomal recessive disorders giving rise to ectropion and keratoconjunctivitis mainly due to exposure.19 This can be so severe as to lead to corneal scarring, infection, and perforation.20 Epidermolytic hyperkeratosis and erythrokeratoderma variabilis are two autosomal dominant varieties. Ichthyosis occurs in the Sjögren-Larsson syndrome, Netherton’s syndrome (ichthyosis, sparse hair, eyebrows, and eyelashes, and atopic diathesis), Refsum’s disease, chondrodysplasia punctata, IBIDS syndrome (ichthyosis, brittle hair, impaired intelligence, decreased fertility, and short stature), and the KID syndrome of ichthyosis, deafness, and keratitis21 (Fig. 33.4B). The keratitis of KID syndrome is attributed to ocular surface drying due to obstruction of the lacrimal gland ductules by shed hyperkeratinized corneal epithelial cells or alternatively to limbal stem cell deficiency.22 Treatment with lubrication, topical corticosteroids, and antibiotics have been advocated with variable success.
Autoimmune blistering diseases
These disorders are developed from the inflammatory reaction of autoantibodies against specific immunoreactants in the skin and mucous membranes. Ocular involvement includes the skin, conjunctiva, and cornea. Corneal opacity results from ocular surface damage due to dry eye − a result of conjunctivitis, scarring of the lacrimal ductules, and loss of goblet cells − exposure due to cicatricial lid change, trichiasis, and infection.23
Erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis
Erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis (see Chapter 15) are self-limited eruptions of the skin and mucous membranes that are due to a hypersensitivity reaction to environmental factors including drugs and infections.24 Since the long-term ocular surface compromise and corneal scarring are considered to be a consequence of the severity of the acute event, initial management is directed toward attenuating local inflammation. Amniotic membrane to the eyelid margins, palpebral conjunctiva, bulbar conjunctiva, and cornea is sometimes successsful.25
Epidermolysis bullosa
Epidermolysis bullosa is an autosomal recessive disorder caused by mutations in the collagen VII gene that compromises synthesis of collagen VII and anchorage fibrils. This results in generalized blistering of the skin and mucous membranes, beginning in childhood. Ocular complications include eyelid blisters, ectropion, symblepharon formation, tear film abnormalities, goblet cell loss, squamous metaplasia, reduced corneal sensation, recurrent corneal erosions (Fig. 33.5), ulceration, limbal stem cell deficiency, corneal pannus, and scarring. Treatment is challenging with some success using amniotic membrane transplantation and autologous transplantation of cultivated limbal epithelial cells on amniotic membrane.26,27
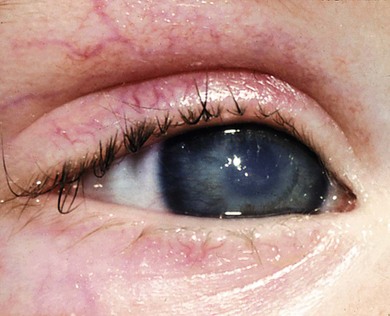
Reiter’s syndrome
Reiter’s syndrome (reactive arthritis) characterized by inflammatory arthritis of large joints, urethritis, and ocular inflammation is commonly associated with conjunctivitis and iridocyclitis. Mainly peripheral anterior stromal keratitis, epithelial erosions, and more diffuse subepithelial and anterior stromal infiltrates associated with fine conjunctival papillae occur. These lesions resolve with topical corticosteroids.28
Vitamin A deficiency and measles (see Chapter 31)
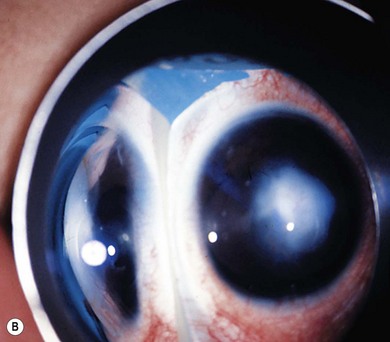
In patients with long-term vitamin A deficiency, the ocular surface can become involved with conjunctival and corneal xerosis, keratinization, severe punctate keratitis, vascularization, and edema. When vitamin A deficiency is accompanied by malnourishment and protein deficiency, keratomalacia, an acute liquefactive necrosis of the cornea can occur (Fig. 33.6), especially when associated with measles infection, herpes simplex, or the use of traditional eye medicines.29 When diagnosed early, some of these problems are reversible with vitamin A replacement. It may be prevented by diet, vitamin A replacement, and measles vaccination.30 Higher doses of vitamin A are necessary when the child has worm infestation or diarrhea.31 Bitot’s spot is a triangular foamy-appearing lesion of keratinized tissue that occurs over the conjunctiva in vitamin A deficiency; its presence on the temporal side of the eye suggests active deficiency.32 Vitamin A deficiency also causes night-blindness.
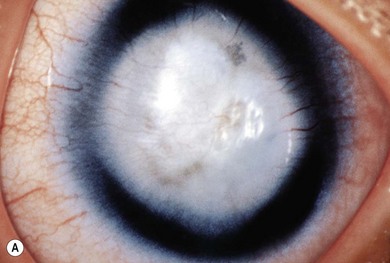
Infectious keratitis (see Chapter 15)
Infectious keratitis is one of the most vision-threatening consequences of trauma, trachoma, and vitamin A deficiency and related ocular surface disease. It becomes the common denominator leading to blindness associated with numerous ocular and systemic conditions. Traumatic corneal injury is a leading predisposing factor,33,34,35,36 but the nature of the trauma and the microbiology of subsequent infection varies based on geography. Recently, corneal trauma due to contact lens wear including orthokeratology has emerged as a distinct risk in regions such as Asia with a high prevalence of pediatric contact lens wear.37–39
Infections following traumatic injury are most commonly caused by bacteria, but a relatively high rate of fungal infection occurs in areas where filamentous fungal corneal infection is more common, such as South India and China.33,36 Whether the leading risk factor is traumatic injury or contact lens wear, the most common organisms include both Gram-positive species (staphylococcal and streptococcal species)33,34,35,37 and Gram-negative species.36,38
Infectious keratitis associated with orthokeratology carries an elevated risk for infection with destructive organisms such as Acanthamoeba.39 Aggressive topical treatment with the appropriate antimicrobial agent offers the best chance of a good optical outcome, but amblyopia due to residual corneal scarring and irregular astigmatism after this unilateral condition can have a significant impact on vision.
Herpetic keratitis (see Chapter 15)
Herpetic keratitis is important in the differential diagnosis of corneal epithelial, stromal, or endothelial disease in childhood. Few case series have been reported. Herpetic keratitis in children may differ from that in adults:40
1. A higher incidence of bilateral disease (in older individuals this is more commonly associated with atopy).
2. A more intense inflammatory response.
3. A propensity toward stromal scarring.
4. Isolated epithelial keratitis, combined epithelial and stromal keratitis, and keratouveitis. Stromal keratitis is most commonly accompanied by epithelial disease.
Inflammatory keratitis: chronic blepharokeratoconjunctivitis (see Chapter 15)
Blepharokeratoconjunctivitis is eyelid margin disease with secondary conjunctival and corneal involvement characterized by inflamed eyelids, anterior lid margin telangiectasia, scaling at the base of the cilia, and chronic papillary conjunctivitis. Corneal findings include punctate epitheliopathy, marginal infiltrates, pannus formation, neovascularization, stromal scarring, and lipid keratopathy.41,42,43
It is recognized in childhood, but standard treatment regimens that include daily lid hygiene, topical broad spectrum antibiotics, systemic antibiotics, and topical corticosteroids must be modified for children. Systemic antibiotics should be restricted to those appropriate for children such as azithromycin. Care must be taken to manage the potential for intraocular pressure elevation that accompanies chronic topical corticosteroid use, particularly since blepharokeratoconjunctivitis in childhood follows a chronic relapsing course. Amblyopia should be considered in patients with axial stromal scarring.44
Interstitial keratitis
Interstitial keratitis describes stromal inflammation, characterized by white cell infiltration and edema that can be followed by neovascularization in prenatal syphilis, mumps, herpes simplex, herpes zoster, varicella vaccination, Epstein-Barr virus infection, leprosy, onchocerciasis, and tuberculosis.45
Congenital syphilitic keratitis is typically a bilateral interstitial keratitis that appears between 5 and 15 years of age, and is characterized by a period of intense photophobia, anterior uveitis, and active, deep stromal infiltration and neovascularization followed by regression and resolution over the course of many months to years leaving typical “ghost vessels.”46 The pathogenesis is not well defined.47 An antigen–antibody complex may be responsible because very few or no spirochetes have been identified in eyes with active keratitis, inflammation does not respond to penicillin and improves with corticosteroids, and antibodies against treponemal antigens have been identified in the cornea.48
Cogan’s syndrome
Cogan’s syndrome consists of interstitial keratitis and audiovestibular disease.49 Commonly a disease of adulthood, Cogan’s syndrome has been recognized in children.50 The keratitis is usually bilateral, peripheral subepithelial stromal inflammation that can progress to nummular lesions. Deep stromal keratitis can lead to deep stromal neovascularization.The eighth nerve impairment may precede or follow corneal involvement. An association with polyarteritis nodosa and other systemic associations has been described.51 The cause is unknown although immunologic factors,52 viral agents,53 and vasculitis54 have been implicated. In atypical disease, uveitis replaces corneal inflammation,55 but the anterior segment inflammation responds well to topical corticosteroids. Although sometimes reversible, hearing loss56 is more commonly persistent; cochlear implants may provide successful rehabilitation.57
Corneal trauma
Exposure keratitis
Exposure keratitis is damage to the ocular surface resulting from inadequate lid protection and failure to maintain adequate lubrication and protection of the corneal epithelium. This results in epitheliopathy, subepithelial and anterior stromal scarring, and, later, corneal neovascularization (Fig. 33.7). In severe cases, often associated with infection, persistent epithelial defect, stromal thinning, and corneal perforation can occur.
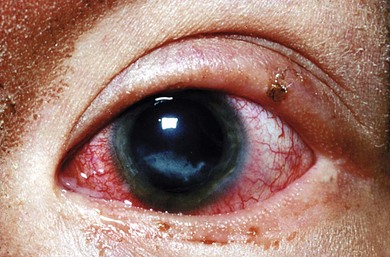
Eyelid defects (colobomas, etc.) may cause exposure keratitis.58 Exophthalmos from orbit disease or craniofacial abnormalities (see Chapter 28) can result in poor lid closure, as can seventh nerve palsies (Fig. 33.8). Fifth cranial nerve palsies also result in neurotrophic epitheliopathy; combined fifth and seventh cranial nerve palsies present the greatest therapeutic challenge. Immediate management requires protection of the eye with ointment and lubrication, but surgical intervention, usually tarsorrhaphy, to establish better eyelid protection of the ocular surface may be required.
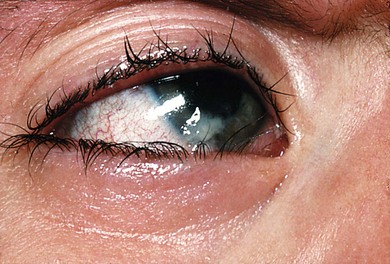
Corneal anesthesia and hypoesthesia
Defective corneal sensation may give rise to a chronic, recurrent, and severe keratitis. Although termed neurotropic, the main etiologic factors are drying, reduced blinking, and repeated trivial trauma. Defective corneal sensation may arise from any cause of fifth nerve damage. It occurs with trauma, herpes zoster ophthalmicus, developmental or acquired brain stem lesions, and tumors, in particular cerebellopontine angle or pontine tumors. It may occur with herpes simplex keratitis.59 Corneal hypoesthesia occurs in leprosy, Goldenhar’s syndrome,60 and other oculofacial syndromes.61 It has been reported in a family of Navajo Indians with an acromutilating neuropathy.62 It can be found in a subclinical form in Adie’s pupil,63 and is characteristic in the Riley-Day syndrome (Fig. 33.9). It occurs in the MURCS association − Mullerian duct aplasia/hypoplasia, renal agenesis or ectopy, and cervicothoracic somite dysplasia.64