Cornea and Systemic Diseases
Farnaz Memarzadeh
Samuel C. Yiu
Ronald E. Smith
Pathologic changes in the cornea are associated with a wide variety of systemic diseases. In some instances, these changes are characteristic of the underlying disease process. Some corneal findings may be secondary to problems of the eyelids, eyelashes, and other adnexal structures that are involved in a systemic disorder. Traditionally, discussions of corneal involvement in systemic diseases have comprehensively categorized the various corneal findings associated with particular diseases. From a practical clinical standpoint, however, patients who consult an ophthalmologist do not usually have a clear-cut systemic diagnosis of their eye complaints. On the contrary, a patient usually presents because of a particular eye symptom, or for a routine examination. At that time, corneal changes may be recognized. It is, therefore, important for the clinician to be aware of the significance of certain corneal changes and their possible relationship to systemic disease processes. At the time of the ophthalmologist’s examination, certain systemic diseases may be subclinical and patients and their internists may be unaware of them, so the familiarity of the ophthalmologist with suspicious corneal findings is of substantial value. In other cases, patients may be referred with a diagnosed or suspected systemic disease, making the association between corneal change and systemic disease more apparent.
The first part of this chapter is devoted to a problem-oriented approach to the evaluation of corneal changes related to underlying systemic diseases. The remainder of the chapter is a more elaborate discussion of the corneal changes associated with systemic disorders, especially inborn errors of metabolism. Systemic and ophthalmologic findings other than those related to the cornea will not be presented in great detail, and the reader is referred to other sections in these volumes for this information.
Problem-Solving
Corneal changes related to underlying systemic diseases are summarized in Table 1. These are categorized as (a) changes that are practically diagnostic or pathognomonic of a systemic disease, (b) changes that are highly suggestive of a systemic disease or group of diseases, and (c) changes that are relatively nonspecific but possibly associated with underlying systemic disorders.
TABLE 15-1. Corneal and Systemic Disease | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The ophthalmologist’s first task is to characterize and identify the clinical problem (e.g., Is the problem a corneal deposit? A peripheral corneal melting ulcer? Is it epithelial? Stromal?). After appropriate classification of the corneal change, Table 1 can be used as a guide to underlying systemic disorders that may be related primarily or secondarily to the corneal finding.
Very few systemic diseases have a pathognomonic corneal manifestation. The deep peripheral corneal ring of Wilson’s disease, the corneal clouding in the mucopolysaccharidoses, and the whorl epithelial dystrophy in Fabry’s disease are almost unique in this regard. However, certain corneal changes are sufficiently characteristic to alert the clinician to the possibility of a type or group of underlying systemic disorders. For example, if the clinical problem is a marginal corneal melting (ulcer or furrow), this might suggest underlying collagen vascular disease, such as rheumatoid arthritis or periarteritis nodosa. Table 1 is designed to suggest to the clinician a particular group of systemic disorders that may be related to corneal findings after a careful determination of the nature of the corneal change (e.g., level of corneal lesion or morphology). Reference to this table may be helpful in delineating both the corneal and systemic diagnoses.
Corneal Changes in Inborn Errors of Metabolism
Disorders of Protein and Amino Acid Metabolism
In the majority of diseases affecting the cornea that are caused by inborn errors of amino acid metabolism, the corneal change is unique and characteristic enough to form the basis for diagnosis. This section describes metabolic diseases related to enzyme blocks in the oxidation of phenylalanine and tyrosine (Fig. 1). Other disorders, such as Wilson’s disease and the paraproteinemias, are also discussed.
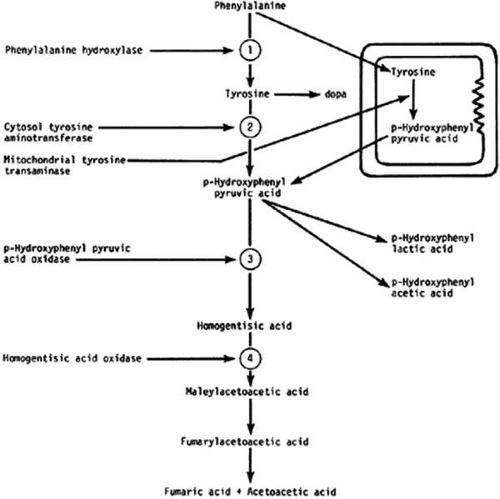 FIGURE 1. Metabolic blocks in the oxidation of phenylalanine and tyrosine. 1, Block of phenylketonuria; 2 and 3, block in tyrosinemia; 4, block in alkaptonuria. |
Alkaptonuria (Ochronosis)
Alkaptonuria is a rare, autosomal-recessive metabolic disease in which the enzyme homogentisic acid 1, 2-dioxygenase is missing. This enzyme is necessary for the oxidation of phenylalanine and tyrosine. As a result of this defect, homogentisic acid, which is normally produced during the metabolism of these two amino acids, cannot be further metabolized and accumulates in the body. The excess metabolite is excreted in the urine, and once oxidized, it turns the urine dark, a feature termed alkaptonuria.
The oxidized by-products of homogentisic acid selectively bind to connective tissue. These tissues become weak and brittle, developing cracks and chips and leading to chronic inflammation and degeneration.1 The most common presentations include dark pigmentation of the pinna of the ear and nasal ala, lower back pain and stiffness as a result of narrowing of joint spaces, hip and knee pain, cardiac valve calcification, and renal and prostatic stones.2,3
Ocular Findings
Ocular involvement is seen in 79% of patients with ochronosis (tissue pigmentation.4 The findings are generally not associated with symptoms and do not cause visual impairment. Ochronotic pigment is found in the sclera (anterior to the insertions of horizontal rectus muscles) and the conjunctiva and at the limbus in the interpalpebral region.4,5,6 Subepithithelial brown particles may be seen in the superficial stroma near the limbus. Pigmentation of the tarsal plates and eyelids has also been described.
Light microscopy reveals the amber globules or fiber-like structures in association with degenerated collagen in the cornea, conjunctiva, episclera, and sclera.7 Ultrastructurally, most of the pigment granules are extracellular, partly altering the collagen fibers and fibrocytes. The ultrastructure of the ochronotic pigment is similar to that of melanin, but histochemically its behavior resembles that of elastin.
Management
There is currently no preventative treatment for the systemic complications of alkaptonuria. The ideal form of therapy would be the replacement of the missing enzyme, which is currently not possible. Other measures, such as dietary restriction of tyrosine and phenylalanine and increasing vitamin C intake, have been attempted. The clinical effect of these treatments has been minimal.
Tyrosinemia Type II (Richner-Hanhart Syndrome)
The tyrosinemias are a group of autosomal recessive disorders of tyrosine metabolism. They are classified as types I-III and neonatal tyrosinemia, which is a transient tyrosinemia occurring most frequently in premature infants.8 Type I tyrosinemia results from the deficiency of fumarylacetoacetate hydroxylase activity and manifests as hepatosplenomegaly, fever, diarrhea, generalized aminoaciduria and neurologic disease early in life.9 There are no skin or eye lesions characteristic of tyrosinemia type I. Tyrosinemia type III results from a primary deficiency of the enzyme 4 hydroxy phenylpyruvate dioxygenase and can vary in presentation from asymptomatic to severe mental retardation and neurologic abnormalities.9
Tyrosinemia type II or Richner-Hanhart syndrome is an oculocutaneous syndrome that results from a genetic deficiency of hepatic tyrosine aminotransferase, the rate-limiting enzyme of tyrosine metabolism. The gene has been localized to chromosome 16q22.10
There is a wide variability in the onset of ocular and cutaneous symptoms, but both usually present in the neonatal period. Skin findings are generally limited to the palms and soles and may be preceded by or coexistent with the ocular lesions. They may start as painful erosive blisterlike lesions that crust and become hyperkeratotic.11 Mental retardation is a variable feature; intelligence may range from normal to severe retardation.
Ocular Findings
Ocular symptoms include epiphora, photophobia and blepharospasm. Ocular signs include corneal haze, pseudodendritic corneal lesions, dendritiform ulcers and rarely whitish corneal and conjunctival plaques.12,13,14 The dendritiform corneal lesions are usually bilateral and stain poorly with fluorescein. Cultures are typically negative. Ocular symptoms may undergo spontaneous remissions and recurrences. Many patients are often misdiagnosed and treated for herpes simplex keratitis. Distinguishing features of tyrosinemia, even in the absence of cutaneous findings, include the usual bilateral presentation, lack of terminal bulbs, minimal staining with fluorescein and rose bengal and lack of response to topical antiviral therapy.12,14,15
A current pathogenetic hypothesis involves crystal production in the cornea as a result of a supersaturated state. When crystal formation is initiated in the central cornea, the disruption of the cells form biomicroscopically evident snowflakelike lesions. The crystals exert a force powerful enough to pierce cell membranes and displace nuclei. Lysosomal enzyme release, polymorphonuclear migration, vascularization, and subsequent healing16 follow cell rupture.
Diagnosis
The combination of skin lesions and pseudodendritic corneal ulcers is almost pathognomonic for this disorder. An elevated plasma and or urine tyrosine level accompanying the typical clinical signs and symptoms is generally adequate for establishing the diagnosis.
Management
A low-tyrosine, low-phenylalanine diet is essential in the management of these patients. The diet usually results in a decrease of the serum and urine tyrosine levels and in relief of the discomfort in the eyes and skin within 24 hours.17 The photophobia and keratitis usually subside within 2 to 4 weeks.
The need for a metabolic examination (including assessment of serum tyrosine levels) in a young child with photophobia and bilateral pseudodendritic keratitis, even in the absence of cutaneous or developmental abnormalities, cannot be overemphasized. If the diagnosis is made early, dietary restrictions of phenylalanine and tyrosine lead to the resolution of ocular and cutaneous changes, and mental retardation may be prevented.18 Penetrating keratoplasty can be performed for end-stage corneal scarring and vascularization. Systemic steroids should be avoided after keratoplasty because dendritic lesions may recur on the new graft.19
Cystinosis
Cystinosis is a rare autosomal recessive disorder with an estimated incidence of approximately 1 in 100,000 to 200,000 live births.20 The gene for cystinosis has been localized to chromosome17p13.21
Cystinosis is a lysosomal storage disease that results from the impaired transport of cystine (a normal product of protein metabolism) from the lysosomes into the cytoplasm.20 Cystine thus accumulates in many internal organs including the kidneys, eye, bone marrow and lymph nodes.22 The mechanism by which the accumulation of cystine causes cellular damage is currently not understood. Recent data suggest that lysosomal cystine increases the rate of apoptosis in cultured cells,23 and a similar process may be at work in vivo.
Three clinical variants of cystinosis have been described. The classic nephropathic cystinosis is the most common, accounting for approximately 95% of the cases reported in North America.20 Initial sysmptoms of cystinosis result from the failure of the renal tubules to reabsorb small molecules causing Fanconi’s syndrome. Symptoms begin in infancy and include polyuria, growth retardation, rickets, and progressive renal failure. In the past, renal failure has usually led to death before puberty, but dialysis and kidney transplantation now allow some patients to reach adulthood. The ocular manifestations include photophobia, cystine deposition in the cornea and conjunctiva, and a peripheral retinopathy.
The intermediate cystinosis or the “late onset” or “juvenile” cystinosis has the same features as the infantile form but with a markedly slower rate of progression.20,24 The adolescent form is characterized by a less severe nephropathy than is seen in the infantile form. Symptoms, which appear in the second decade, may or may not include rickets, renal failure, or the characteristic corneal and conjunctival deposits. Retinopathy is absent. Life expectancy is decreased because of renal dysfunction. The mode of inheritance is autosomal recessive.
The ocular, or non-nephropathic form, formerly called “benign” or “adult” form is asymptomatic, with the possible exception of photophobia, and is usually diagnosed during a routine slit-lamp examination.25,26 Renal function is normal and patients have a normal life expectancy.
Ocular Findings
The outstanding clinical feature common to all three phenotypes is the corneal and conjunctival cystine crystal deposition (Figs. 2 and 3). Photophobia is often the only presenting visual symptom; this may be incapacitating and associated with blepharospasm.
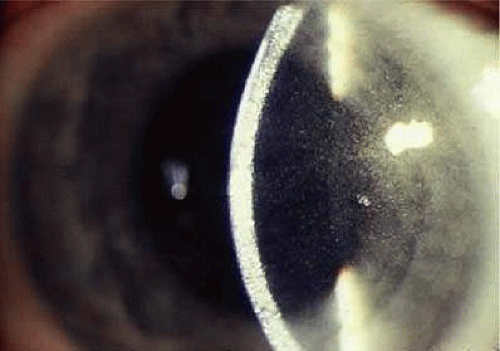 FIGURE 2. Cystinosis. Refractile crystals noted throughout the corneal stroma. |
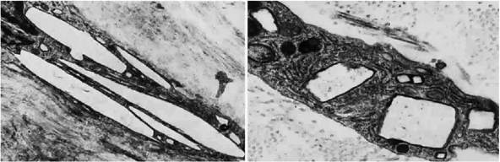 FIGURE 3. Cystinosis. Top, Transmission electron micrograph showing a stromal keratocyte containing a number of needle-shaped crystalline profiles limited by unit membranes. (19,000 ×) (top). Electron micrograph of conjunctival fibrocyte revealing crystalline profiles of sparse granular material within membrane-limited lysosomes (30,000 ×) (bottom). (Courtesy of Dr. Kenneth Kenyon), |
Corneal deposits appear as a layer of homogeneously distributed, fusiform or needle-shaped, iridescent crystals situated in the stroma beneath the epithelium. In the infantile form, anterior crystal deposition begins early in life (between 6 and 15 months of age) and proceeds posteriorly as the patient ages; deposition advances more rapidly in the periphery. The anterior location of the crystals may be associated with recurrent erosions.27 The depth of the stromal deposition and the density of crystals is always greater peripherally than centrally. More and larger crystals occur in the superficial stroma. No visual impairment occurs at this early stage. By the age of 7 years, most patients have crystals, either within or on the endothelial surface.28,29 Markedly decreased corneal sensitivity is also present.30 The spherical contrast sensitivity function is significantly lower in infantile cystinosis than in age-matched controls.31
The conjunctiva has a ground-glass appearance. Polychromatic, polymorphic, rectangular, or rhomboidal crystals can be seen on slit-lamp examination.
The uvea contains an abundance of polymorphous crystals. Clinically, these can be seen as glistening dots on the surface of the iris. Thickened iris stroma and posterior synechiae may occur; pupillary block glaucoma32 has also been reported. The entire uvea has polymorphic crystal deposition, most heavily in the choroid. The sclera also has crystal deposition.
The retinal abnormality consists of a generalized depigmentation that may assume a patchy pattern. At first the pigmentary disturbance tends to be peripheral, but it progresses with age. Macular abnormalities have been observed.33 Intracellular crystals have been seen in the retinal pigment epithelial cells during electron microscopy.
Diagnosis
The ocular findings of cystinosis are sufficiently unique and characteristic to form the basis for a diagnosis. The diagnosis can also be made by measuring the cystine content of polymorphonuclear leukocytes.20 Conjunctival biopsy has also been used for diagnosis.34 Prenatal diagnostic testing is available for at-risk families.35
Differential Diagnosis
Polychromatic corneal crystals similar to those in cystinosis also may be seen in multiple myeloma,36 Schnyder’s crystalline dystrophy, Bietti’s crystalline dystrophy, gout, and chrysiasis.
Management
Oral cysteamine is the only available treatment for the nephropathy of infantile cystinosis. The mechanism of lysosomal cystine depletion involves the entry of cysteamine into the lysosome and reaction with cystine to form the cysteamine-cysteine compound which can exit through an intact lysine transporter.37 The cystine depletion that occurs apparently helps stabilize glomerular function and also improves growth and development.38 Cysteamine treatment decreases the deposition of additional cystine in the kidneys; but it does not reverse existing renal tubular and glomerular damage or gradual loss of kidney function.39
Renal transplantation is an alternative therapy for patients with advanced renal disease. As a result of renal transplantation, children are now surviving to the second and third decades with normal renal function. Although cystine is not deposited in a grafted kidney, it appears to accumulate relentlessly in other organs and tissues, especially ocular tissue, and progressive visual impairment has been documented.40 Continued therapy with oral cysteamine after renal transplantion can be useful in preventing such complications.20
There is no evidence that corneal cystine deposition or rod and cone dysfunction are reversible with cysteamine treatment; it may only prevent further ocular damage. Reversal of corneal crystal deposition by topically administered cysteamine has been reported.41,42 Ocular sysmptoms improve within a period of weeks and the corneas clear within months. Although penetrating keratoplasty can be performed for advanced corneal cystinosis with endothelial decompensation, cystinosis can recur in the transplanted graft.27
Wilson’s Disease (Hepatolenticular Degeneration)
Wilson’s disease is an autosomal recessive inherited disorder of copper metabolisom that results in pathological accumulation of copper in many organs and tissues. The genetic defect has been mapped to chromosome 13q.43 It is suggested that free radical formation and oxidative damage, possibly mediated via mitochondrial copper accumulation, are important in the pathogenesis of the disease. Accumulation of copper within hepatic mitochondria leads to premature oxidative damage. Further damage of the hepatocytes and ensuing inflammation and fibrosis is caused by the release of copper from necrotic hepatocytes.44,45,46
Clinical manifestations rarely occur before 6 years of age and may be delayed until the fifth decade. Liver disease and neuropsychiatric disturbances are the most common presentations.45 Liver involvement can range from asymptomatic elevation of liver enzymes, to fulminant hepatic failure and cirrhosis. The leading neurologic symptoms are dysarthria, dyspraxia, ataxia and progressive extrapyramidal signs.45 Psychiatric manifestations, such as depression, labile mood or psychosis, are also common.
Ocular Findings
One of the few truly diagnostic and pathognomonic physical signs in clinical medicine is a Kayser-Fleischer corneal pigment ring found in patients with Wilson’s disease (Fig. 4). This ring is recognized as a golden-brown, ruby-red, or green band of 1.0 to 3.0 mm, starting at the limbus but at the level of Descemet’s membrane.47 The color of the ring is presumably caused by scattering and reflection of incident light and by photointerference effects created by the layers of copper granules. Such variables as size, shape, and unit density of the granules may account for the different appearances of the Kayser-Fleischer ring. The course of the Kayser-Fleischer ring has been well documented.48,49 The site of earliest pigment deposition is an arc in the superior periphery of the cornea from the 10- to 2-o’clock meridian. The arc spreads slowly toward the horizontal plane and gradually broadens. Later in the progression of the ring formation, a band appears inferiorly as a crescent stretching from the 5- to 7-o’clock positions. In time, the two arcs meet. With treatment, the sequence of events is reversed, and after the copper has reabsorbed, a pitted or beaten silver pattern may become apparent at the previous site of the ring. Decreased visual acuity is not an associated feature.
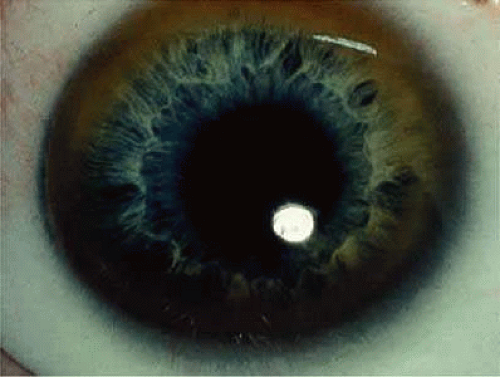 FIGURE 4. Wilson’s disease. Kayser-Fleischer peripheral corneal ring. Copper pigment at level of Descemet’s membrane. |
A rare ocular manifestation of Wilson’s disease is the “sunflower cataract”. These cataracts result from copper deposition in the anterior and posterior capsule and can reduce visual acuity.50
Diagnosis
This syndrome should be suspected clinically. A low-serum ceruloplasmin level along with the presence of Kayser-Fleischer rings and the typical neurological symptoms establishes the diagnosis.51 Other positive test results can include a high 24-hour urinary copper excretion, elevated serum “free” copper and an elevated hepatic copper level as shown by liver biopsy.
Differential Diagnosis
Pigmented corneal rings clinically identical to Kayser-Fleischer rings have been reported in non-Wilsonian liver diseases, including primary biliary cirrhosis, progressive intrahepatic cholestasis of childhood, and chronic active hepatitis. Although these diseases cause an elevated level of copper in the blood, urine, and liver, only in Wilson’s disease are subnormal levels of ceruloplasmin present.52
Management
The mainstay of treatment for Wilson’s disease is pharmacologic therapy. Treatment for symptomatic patients should include a chelating agent (penicillamine or trientine) that mobilizes copper and increases its excretion in the urine.43,52 Treatment of presymptomatic patients or maintenance therapy of successfully treated symptomatic patients can be accomplished with the chelating agents or with zinc.53 Zinc interferes with the intestinal absorption of copper. Liver transplantation, which corrects the underlying hepatic defect in Wilson’s disease, is the treatment of choice in patients with fulminant disease or in those with cirrhosis. With pharmacologic therapy or after liver transplantation, the Kayser-Fleischer rings regress.54,55
Lowe’s Oculocerebrorenal Syndrome
This syndrome is an X-linked recessive multisystem disorder that involves the eyes, nervous system and kidneys. It is characterized by mental retardation, abnormalities of the musculoskeletal system, Fanconi syndrome of the proximal renal tubules with aminoaciduria and congenital cataracts and glaucoma. The syndrome is caused by a mutation of the OCRL-1 gene localized to the Xq25 region.56
Ocular Findings
Congenital cataracts are present in all patients with Lowe’s syndrome and are related to lenticular dysmorphogenesis, which can be recognized in utero.57 The cataract has a characteristic dense, small discoid shape. Degeneration of the posterior lens fibers and defects in the lens capsule have been identified.58 Glaucoma is present in up to 64% of patients.59 This syndrome is typically recognized in the first year of life and is associated with buphthalmos, corneal enlargement and clouding, tearing and photophobia.60,61 The angle anomaly of patients with Lowe’s syndrome is thought to resemble that seen in primary congenital glaucoma.61,62 It is believed that the secondary aphakic glaucoma associated with cataract extraction during the first year of life is also responsible for development of glaucoma in these patient.61
Diagnosis
This disorder should be considered in any male infant with both congenital cataracts and congenital glaucoma. In carrier females, the lenticular opacities are punctate, white to gray in hue, and vary in size from microns to several millimeters. Unlike congenital nuclear opacities, lenticular opacities are present in all layers of the cortex, indicating that they are formed well into adult life. Subcapsular, plaquelike cataracts are seen predominantly in older obligate carriers. Female carriers have an identifiably higher number of lens opacities than do age-matched controls; therefore, genetic counseling may be indicated since 50% of their female offspring are expected to be carriers.63
Biochemical findings are typical and include metabolic acidosis and renal tubular acidosis. The basic enzyme deficiency is unknown but may be related to biochemical abnormalities in glycosaminoglycan metabolism. The urinary findings include proteinuria, generalized aminoaciduria, organic aciduria, and on occasion, glycosuria.64
Differential Diagnosis
Rubella can also cause both congenital cataracts and glaucoma.
Management
Early identification and surgical removal of cataract by lensectomy-vitrectomy is recommended for patients with dense cataracts. These patients should be monitored closely for changes in IOP, optic nerve cupping and refractive error. Despite timely and appropriate treatment, visual acuity results are rarely better than 20/70 and nystagmus is likely.59
Porphyria
Porphyrias are a group of rare metabolic disorders characterized by the accumulation of photosensitive and toxic intermediates of the heme metabolism pathway in various organs of the body, including the skin, liver and eye. These disorders are classified as either hepatic or erythropoietic, depending on the primary site of overproduction and accumulation of the porphyrins, but there are overlapping features. The major features of the hepatic pophyrias are neurologic, including abdominal pain, neuropathy and mental disturbances, whereas erythropoietic porphyrias typically cause cutaneous sensitivity.65 Specific systemic and ocular manifestations of two of the porphyrias are discussed below.
Congenital Erythropoietic Porphyria
This autosomal-recessive disorder is also known as Gunther’s disease. It is caused by a deficiency of uroporphyrinogen III synthase activity, resulting in an overproduction of porphyrins (type I) for which the body has no use. Different molecular defects have been detected.66 The clinical signs include hydroa aestivate (a vesicular or bullous eruption of the face, backs of the hands, and other exposed areas of the body), erythrodontia (brownish discoloration of the teeth), hypertrichosis, and splenomegaly.67 Neurologic symptoms, hypertension, and abdominal colic are not found.
Ocular Findings
Ocular manifestations of this disorder include cicatricial ectropion, loss of eyelashes and eyebrows, bilateral exophthalmos, optic atrophy, retinal hemorrhages, and chorioretinitis.68,69 Conjunctivitis may result in marked scarring and adhesion of the conjunctiva to the sclera. Decreased corneal sensitivity, corneal scarring and vascularization, corneal perforation at the limbus, and scleromalacia may occur. Histopathology of tissue from a patient who underwent a penetrating keratoplasty and conjunctival resection revealed a thickened basement membrane of the vessels within the conjunctival and corneal stroma. Microfibrillar material was seen in the extracellular spaces of the conjunctival stroma. Inflammatory cells were noted in the corneal stroma, and there was a loss of keratocytes. Descemet’s membrane lacked the normal fetal and postnatal banding and the endothelium was severely damaged.70 Changes in the vessels and conjunctival stroma were similar to those described in the skin of porphyria patients exposed to ultraviolet light.
Diagnosis
Normochromic anemia and excessive excretion of uroporphyrin I and coproporphyrin I in urine and feces are found. The diagnosis should be confirmed by detection of decreased uroporphyrinogen III synthase activity.
Management
Protection from sunlight is important. Oral β-carotene may improve tolerance to sunlight. Transfusion of sufficient blood to suppress erythropoiesis is effective but results in iron overload.65
Variegate Porphyria
This hepatic porphyria is characterized by acute attacks involving abdominal and neurologic manifestations, as well as by chronic skin lesions that are caused by photosensitivity.
Ocular Findings
In addition to the previously mentioned ocular signs, optic neuritis, optic atrophy, and exudates similar to those in acute intermittent porphyria are seen in affected homozygotes.69
Management
Intravenous heme leads to a rapid recovery of acute attacks. No specific therapy is available for ocular lesions. Avoidance of sun exposure is effective in treating skin lesions. Scleral patch grafts have been used successfully to treat the scleromalacia perforans that develops in some patients.71
Amyloidosis
Amyloid deposition produces dysfunction in any organ by replacing its functioning mesenchymal or parenchymal cells. All amyloid proteins are insoluble fibrinous proteins that share a unique fibrillar ultrastructure. Depending on the origin of the amyloid protein, the amyloid fibrils deposit either in certain organs or systemically. Amyloid deposition does not incite any inflammatory response; however, a foreign body giant cell reaction may be seen occasionally around deposits in the ocular adnexa.72
Many classifications of amyloidosis exist. A widely used clinical classification is based on the extent of involvement of tissues by amyloid (local vs. systemic) and presence or absence of concomitant systemic or local disease (primary vs secondary).
More recently, amyloidosis has been classified according to the protein precursor. The nomenclature for this classification includes A (for amyloidosis) followed by the initial of the protein precursor (e.g., AL for amyloidosis with light chain immunoglobulins as precursors).73 The most common type of systemic amyloidosis currently seen is the light chain amyloidosis (AL), which is associated with primary amyloidosis and sometimes with multiple myeloma. Fifteen to 20% percent of patients with myeloma have amyloidosis; fewer than 20% of patients with amyloidosis have myeloma.74 In secondary systemic amyloidosis the major amyloid deposit is protein AA, a degradation product of a serum acute phase reactant, serum amyloid A (apoSAA).
Histologic examination of amyloid deposition has shown a pericollagenous distribution associated with the primary and myeloma-related types (pattern type I) and perireticulin deposition in the secondary form of amyloidosis, associated with parenchymatous organ invasion (pattern type II).
Localized Primary Amyloidosis
Primary amyloidosis is the form most commonly associated with clinically important depositions of amyloid in the eyes or ocular adnexa.73,75
Two forms of localized inherited corneal involvement may occur. First is lattice corneal dystrophy types I (Biber-Haab-Dimmer) and III which are charachterized by the presence of refractile or so called “lattice” line in the corneal stroma (see corneal dystrophies section) (Fig. 5).76 The second is gelatinous droplike dystrophy. Characteristic lesions of glatinous droplike dystrophy appear as centrally located, raised, gelatinous masses with a mulberry-like surface (Fig. 6).77,78 For a more detailed discussion please see the corneal dystrophy section of this book.
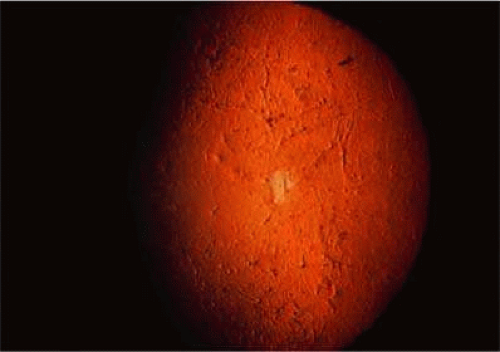 FIGURE 5. Lattice dystrophy. Localized amyloid of the cornea. Corneal changes are shown in retroillumination. |
 FIGURE 6. Rare primary familial amyloidosis. |
Systemic Amyloidosis
Rarely, patients with lattice corneal dystrophy (type II) may have a familial primary systemic amyloidosis known as Meretoja syndrome. These patients develop lattice dystrophy in young adulthood and, in later years, skin changes, cranial nerve palsies, peripheral neuropathies, and visceral complaints. The lattice dystrophy that develops with systemic involvement is milder with regard to visual loss but more extensive in that it reaches to the corneal periphery without the peripheral lucid interval seen with localized lattice.79 Other recorded ocular manifestations include abnormal conjunctival vessels, pupillary abnormalities (decreased light reflex, pupillary deformity and amyloid deposition at the pupillary border), and keratoconjunctivitis sicca.80
Conjunctiva
Clinically detectable conjunctival involvement is not a feature of systemic amyloidosis. However, localized amyloidosis confined to the conjunctiva has been reported as an example of primary amyloidosis. Conjunctival amyloidosis is often asymptomatic and may be present for years before the patient seeks medical attention. Typically, there is a discrete, nonulcerative, yellow, waxy, firm, nontender subconjunctival swelling (Fig. 7). This may be located in the palpebral fornix or bulbar conjunctiva, including the limbal area. The conjunctival area is usually smooth but may be friable and may show recurrent bleeding. However, antecedent local diseases have been incriminated in this amyloid deposition.76
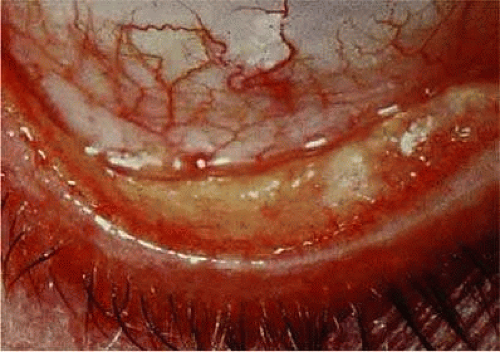 FIGURE 7. Conjunctival amyloidosis. |
Eyelid
The skin of the eyelid is the site of predilection for the characteristic cutaneous eruption of primary systemic amyloidosis.76 Typically, the lesions are symmetric, bilateral, small, smooth, and discrete or confluent papules. These lesions may be yellow and waxy or purple and hemorrhagic. A positive family history is often present. Primary localized amyloidosis does not affect the skin of the eyelids.
Vitreous
Vitreous opacities in amyloidosis may be uniocular or they may be asymmetric if both eyes are involved; these opacities may be the presenting feature of systemic amyloidosis. Vitreous involvement is much more common in primary familial systemic amyloidosis than in nonfamilial amyloidosis.81
Orbit
Orbital involvement does occur in primary localized amyloidosis. Common presenting symptoms include ptosis, proptosis and globe displacement.82 Amyloid also occurs in the orbits of some patients with primary familial systemic amyloidosis.
Neuro-Ophthalmic Manifestations
Pupillary abnormalities are the most common neuro-ophthalmic sign found in primary familial systemic amyloidosis. Pupils have been described as dilated and unequal with absent or sluggish reactions to light, which at times may be accompanied by poor accommodative responses.73,81 Amyloid may be deposited in ciliary nerves. External ophthalmoplegia, diplopia, and optic neuropathy have been reported in some cases of familial amyloidosis.
Secondary Systemic Amyloidosis
Secondary systemic amyloidosis is associated with a number of underlying afflictions that have a significant chronic inflammatory component, such as tuberculosis, osteomyelitis, and leprosy. Small, clinically insignificant deposits of amyloid in the eye have been described rarely, and ophthalmologic signs attributable to the presence of amyloid are virtually unknown.76
Secondary Localized Amyloidosis
This form of amyloidosis usually follows chronic diseases, such as connective tissue diseases, neoplastic disorders or infectious diseases. Corneal involvement has been reported to occur in association with interstitial keratitis, trachoma,83 bullous keratopathy, trichiasis,84 granular corneal dystrophy,85 and keratoconus,86 or after chronic keratitis.
Diagnosis
Amyloidosis must be suspected on clinical grounds. Definitive diagnosis depends on histochemical reactions, including metachromasia with crystal violet, fluorescence with thioflavin T, birefringence and dichroism with congo red, and positive staining with direct cotton dyes, such as sirius red. The mere recognition of amyloid in an ocular structure can no longer be considered a complete diagnosis. In primary systemic amyloidosis, the diagnosis is readily made by pathologic examination of the vitreous aspirate. The possibility of amyloidosis in extraocular sites, underlying or associated ocular disorders, familial aspects, and myelomatosis and other immunoglobulin abnormalities must be considered.
Management
Medical treatment may be specific for the various amyloidoses. Liver transplantation has been successful in some familial cases. Treatment of the underlying inflammatory condition is of questionable benefit in secondary amyloidosis; however, in primary amyloidosis, where a presumed monoclonal plasma cell secretes excess light chain, alkylating agents have been used with modest impact on survival. Currently a combination of high-dose melphalan and autologous stem cell transplant is under investigation and is believed to be a promising future treatment.87 When surgery is indicated, the treatment of choice for vitreous involvement of amyloidosis is vitrectomy. Pars plana vitrectomy can be effective in restoring visual acuity, although recurrences are common. Reopacification of the retrolental vitreous is the most common reason for vitrectomy revision, which is required in 24% of patients.88 Penetrating keratoplasty may be required for advanced corneal disease although deposition frequently recurs in the graft. Phototherapeutic keratectomy with excimer laser has been used for superficial depositions in the cornea.89,90
Gout and Urate Keratopathy
Gout is a clinical disorder of purine metabolism manifested by hyperuricemia, recurrent attacks of acute arthritis that are usually responsive to colchicine, and on occasion, tophaceous deposition of monosodium urate. Nephropathy is a frequent complication.91 Primary gout is usually attributed to an inborn error of metabolism. A deficiency of hypoxanthine-guanine phosphoribosyl transferase has been found in familial gout and may represent the enzymatic deficiency in at least one variety of the disease.92 The largest subgroup, however, consists of patients in whom the biochemical defect is as yet undefined. Other enzymes may be involved.
Secondary gout or hyperuricemia may occur in toxemia of pregnancy and chronic renal insufficiency, as well as in other conditions that may be associated with breakdown of cell nuclei, such as malignant lymphomas, chronic myelogenous leukemia, hemolytic anemia, and primary polycythemia.
Increased endogenous urate production, as well as impaired renal urate excretion, has been cited as the basis for the hyperuricemia. The characteristic pathologic feature of gout is the deposition of urate crystals in articular and periarticular structures. In the eye, precipitation of urate crystals has been described in the lens, iris, anterior chamber, conjunctiva, sclera, tarsal plates, tendons of extraocular muscles, and cornea.93,94,95,96,97,98
Ocular Findings
Clinical manifestations of ocular gout include chronic hyperemic conjunctivitis, scleritis, episcleritis (Fig. 8), elevated intraocular pressure and asteroid hyalosis.99 Monosodium urate crystals may be deposited in the corneal epithelium.97 Slit-lamp examination shows a fine, golden-yellow, scintillating crystal formation, diffusely distributed in the cornea, more heavily in the interpalpebral fissure and extending to the limbus. These crystals appear epithelial and subepithelial; they are best visualized by retroillumination.
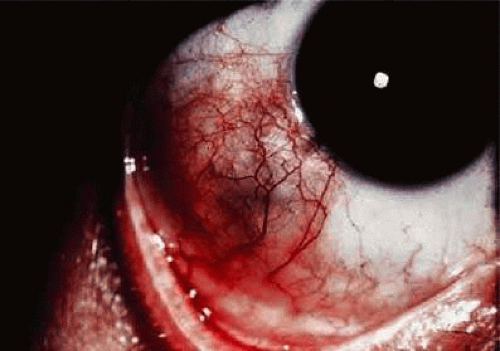 FIGURE 8. Episcleritis in gout. |
Crystals interpreted as urates also have been described in band keratopathy occurring in a patient with gout.100 This keratopathy cannot be differentiated from calcium deposits solely by slit-lamp examination. The site of urate crystals in the corneal epithelium is intranuclear.97 Uratelike crystals also have been seen in the corneal stroma in a disease termed keratitis urica. This is probably a localized dystrophic corneal disease and not associated with gout.101
Diagnosis
A high index of suspicion is necessary to establish gout as a contributing factor to corneal crystals, band keratopathy and conjunctivitis. Accordingly, the ophthalmologist should consider gout in the differential diagnosis of every patient with bilateral chronic conjunctivitis. High levels of uric acid in the urine and blood are helpful in establishing the diagnosis. Diuretic therapy is currently one of the major causes of secondary hyperuremia.101 Other causes of corneal crystals include cystinosis and monoclonal gammopathy. (See Table 1 for the differential diagnoses of crystals and band keratopathy).
Management
Anti-inflammatory agents, such as indomethacin and colchicines, are used to treat acute attacks of gouty arthritis. Allopurinol, a xanthine oxidase inhibitor, is the most commonly used agent for long-term treatment of gout and reduction of serum urate levels. Febuxostat is a newer xanthine oxidase inhibitor and in some studies has been found to be more effective than allopurinol in reducing serum uric acid levels.102 Long-term use of allopurinol has been associated with an increased risk of cataract formation in some studies.103,104
Multiple Myeloma
Multiple myeloma is a malignancy characterized by monoclonal expansion of plasma cells. The neoplastic cells secrete monoclonal immunoglobulins termed light chains (Bence Jones proteins) or heavy chains. The disease usually presents with nonspecific symptoms of fatigue, anorexia and weight loss. Bone involvement with osteolytic lesions and pathologic fractures resulting in significant bone pain is a common presentation.105 Bone marrow infiltration results in pancytopenia. Common laboratory findings include anemia, hypercalcemia, renal dysfunction, Bence Jones proteinuria and a monoclonal globulin spike on serum electrophoresis. The diagnosis can be confirmed by bone marrow biopsy, which shows an uncontrolled proliferation of plasma cells.
Ocular Findings
The ocular manifestations of multiple myeloma may be divided into two groups: those attributable to plasmacytoma growth in and about the eye, and those attributable to hematologic and serum protein abnormalities.106 The ocular manifestations include intraorbital or intraocular plasmacytomas,107,108 ciliary body cysts,109,110 retinal vascular tortuosity, hemorrhages and exudates,108,111 and the ophthalmic manifestations of intracranial plasmacytomas (papilledema).112 Crystals may be seen in the lens, and the cornea may show band keratopathy resulting from the hypercalcemia. Clusters of myeloma cells may occur on the corneal endothelium and in the anterior chamber as a pseudohypopyon.112 Conjunctival involvement in patients with multiple myeloma includes sludging of red cells in conjunctival vessels; occasionally malignant plasmacytomas of the conjunctiva arise anew or in association with advanced multiple myeloma.113
The presence of numerous delicate, scintillating crystals in the cornea has been recognized as a rare manifestation of hypergammaglobulinemia.36,114,115,116 In some cases, the corneal crystals are interspersed throughout the corneal stroma; in others they are most evident in the anterior cornea,117 including the epithelium.118 Posterior stromal corneal deposits are uncommon. The deposits usually are not vascularized, and corneal sensation is normal. Crystals may be detectable clinically in the bulbar conjunctiva.36,116,117 Aronson and Shaw described corneal crystals as probably consisting of cholesterol;36 however, these crystals probably consist of partial or complete immunoglobulins (Fig. 9).119 Cysts of the ciliary body have been reported in 33% to 50% of myeloma patients, and retinal vascular lesions have been reported in up to 66% of these patients.115 Although corneal and orbital involvements are less common, orbital involvement is the first manifestation of systemic disease in about 75% of cases. Primary plasmacytomas may arise in the soft tissues or in the surrounding bones with secondary orbital invasion. The orbital roof and frontal bones often are affected, with resultant proptosis and downward displacement of the globe. The most common clinical signs are eye pain, diplopia, and visual impairment associated with a history of slowly progressive proptosis for weeks to months.108 Choroidal folds, venous engorgement of the disc, ptosis, and pupillary involvement may occur.
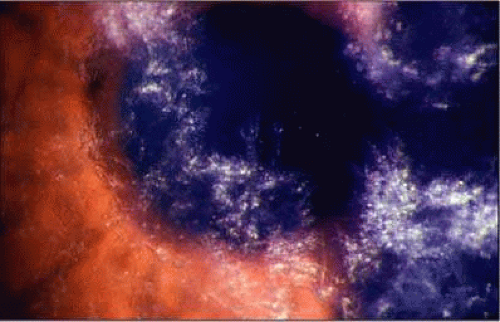 FIGURE 9. Multiple myeloma. (Courtesy of Dr. Dan B. Jones). |
Polychromatic, dustlike deposition of copper in Descemet’s membrane of the central cornea (with peripheral sparing) and in the anterior and posterior lens capsule occurs when the myeloma protein has strong copper-bonding properties.119,120
Diagnosis
Serum protein electrophoresis, examination of the urine for the presence of Bence Jones protein, and bone marrow aspiration are required. The differential diagnoses must include other causes of corneal crystals (Table 1).
Management
The treatment of multiple myeloma is directed toward the systemic disease. Melphalan was the first described treatment for multiple myeloma and is currently the most widely used regimen. Newer therapies include the combination of melphalan with steroids and autologous stem cell transplantion, angiogenesis inhibitors, such as thalidomide and immunotherapy.122 Chemotherapy and radiotherapy are effective in reducing the tumor mass in the orbit. Radiotherapy is effective in treating orbital plasmacytomas and localized lytic bone lesions.
Ciliary body epithelial cysts are usually not diagnosed in life and do not require treatment. Retinopathy may improve with systemic treatment. Both penetrating and lamellar keratoplasties have been performed for advanced corneal disease, but recurrence after lamellar keratoplasty has been reported.119 Marked clearing of the corneal crystals with corresponding improvement of visual acuity has been reported to occur after chemotherapy.123
Disorders of Lipoprotein and Lipid Metabolism
Lipoproteins are macromolecular complexes that carry hydrophobic lipids (cholesterol and triglycerides) in the plasma. Lipoproteins are spherical structures made primarily of lipid and protein. The hydrophobic nonpolar lipids (esterified cholesterol and triglycerides) are located in the core of lipoproteins. The phospholipids and a small quantity of free or unesterified cholesterol, which are more hydrophilic, cover the surface of the particles, allowing miscibility in plasma. A family of proteins, the apolipoproteins, also occupies the surface of lipoproteins and plays a crucial role in the regulation of lipid transport and lipoprotein metabolism.124,125,126
Lipoproteins can be classified on the basis of their size, composition and densities. The lipoproteins become smaller and more dense as they reduce their triglyceride content and increase their protein content. They are classified into five major classes: chylomicrons, very low density lipoproteins (VLDL), intermediate-density lipoproteins (IDL), low-density lipoproteins (LDL), and high-density lipoproteins (HDL).
Hyperlipoproteinemia
Five basic phenotypes (Table 2) of hyperlipoproteinemia have been defined;126,127,128 these five phenotypes are distinguished by the class of lipoprotein that is elevated in each.
TABLE 15-2. Hyperlipoproteinemias | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Type I (Hyperchylomicronemia). A deficiency of lipoprotein-lipase and Apo CII result in an elevation of chylomicrons, which are normally absent from fasting serum.127 Most patients are asymptomatic; however, attacks of abdominal pain, and sometimes pancreatitis, do occur. Eruptive xanthomas of the skin or mucous membranes, tendinous tuberous xanthomas and corneal arcus may be seen. Occasionally, lipemia retinalis is present. Cardiovascular complications are not common. This disorder is transmitted as an autosomal-recessive trait.128,129
Type II (Hyperbetalipoproteinemia and Prebetalipoproteinemia). In these disorders there are elevated levels of low-density lipoproteins (LDLs; type IIa) or elevated levels of both LDLs and very low-density lipoproteins (VLDLs; type IIb).127 The most serious systemic manifestation is cardiovascular disease, including peripheral vascular disease and myocardial infarction. The serum is clear, and there is an elevated cholesterol level. Corneal arcus, xanthelasma, or conjunctival xanthomas are typically present. This disorder is transmitted as an autosomal-dominant trait. Type II is expressed most severely in the homozygous state. Death before the fourth decade is common.128,129
Type III (Dysbetalipoproteinemia). VLDL triglyceride and VLDL cholesterol are both elevated in this rare disorder.127 The serum is creamy. A variety of xanthomas are seen, including palmar, tuberous, and subperiosteal varieties and the more characteristic tuberoeruptive variety. Corneal arcus and lipemia retinalis are present early in the disease. Cardiovascular diseases, including peripheral vascular disease and myocardial infarction, are the most serious systemic manifestations. Type III disease is inherited as an autosomal-recessive trait.128,129
Type IV (Hyperprebetalipoproteinemia). Serum cholesterol in this disease may be normal or slightly elevated; however, the triglycerides are elevated and overproduction of VLDLs has been reported.127 There is no apparent increased risk of coronary heart disease in this type.127 Corneal arcus and xanthelasma are not seen; but lipemia retinalis may be evident. Type IV is transmitted as an autosomal-dominant trait.128,129
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


