Chapter 31 Conjunctiva and subconjunctival tissue
Anatomy
Beneath the conjunctiva lies a fibroelastic tissue, Tenon’s capsule, which surrounds the eye ball from the corneoscleral junction to the optic nerve. Tenon’s capsule is thicker in children and contains more fibroblasts. Hence surgeries like trabeculectomy performed in children, especially without adjuvant procedures like intraoperative use of antimetabolites, may fail due to the aggressive healing response induced by these fibroblasts.1
Conjunctiva in systemic disease
Vitamin A deficiency
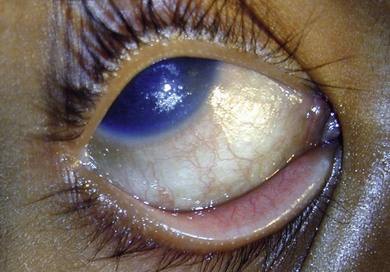
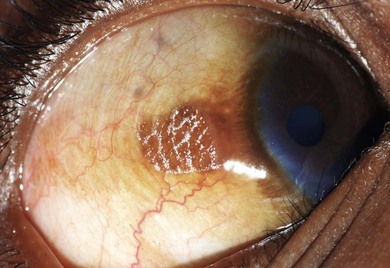
In this condition, the conjunctival epithelium is transformed from the normal columnar to the stratified squamous type. There is an associated loss of goblet cells, formation of a granular cell layer, and keratinization of the surface. The conjunctiva loses its normal lustre and is altered into a dry or unwettable one (Fig. 31.1). It is almost always bilateral. A classic ocular sign is Bitot’s spots, which is a superficial, scaly, gray area on the interpalpebral region of the bulbar conjunctiva (Fig. 31.2). Corynebacterium xerosis can colonize these spots, and produce a foamy appearance because of the gas-forming nature of these organisms. If untreated, the condition involves the cornea, causing corneal xerosis and finally corneal melting, or keratomalacia.
Parenteral administration is indicated in those children with conditions such as persistent vomiting, severe stomatitis, and attendant difficulty in deglutition, severe diarrhea with malabsorption, and septic shock. Such children can be treated with intramuscular injection of 55 mg of water-miscible retinol palmitate (100 000 IU), which replaces the first oral dose. This is repeated the next day. Children of less than 1 year are treated with vitamin A in half the prescribed dosage. After the acute phase is over, dietary supplements with provitamin A-rich foods, should be provided.2
Xeroderma pigmentosa
This condition is inherited as an autosomal recessive disorder. Symptoms appear in early childhood. Affected individuals present with extreme photophobia, photosensitivity, and typical dark pigmentary changes in the skin. They are at an increased risk for malignant lesions in sun-exposed mucocutaneous and ocular structures (Fig. 31.3A). There is an impaired ability to repair ultraviolet light-induced DNA damage, which results in accumulation of the damaged DNA. This accumulation of abnormal DNA leads to chromosomal mutation and cell death and is thought to be responsible for neoplasms in these individuals.
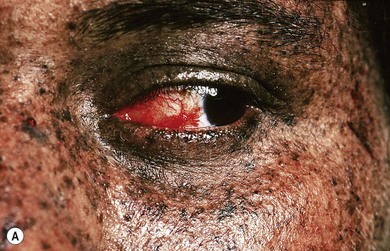
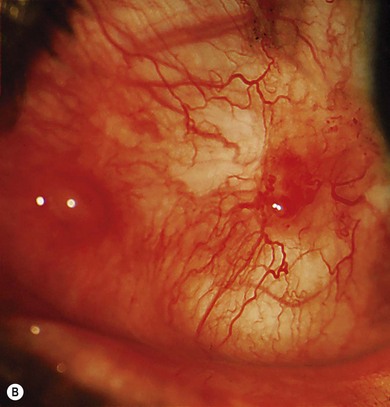
Conjunctival involvement occurs mostly in the interpalpebral area in the form of xerosis, telangiectasia, chronic conjunctival congestion, pigmentation, pinguecula, and pterygium. Ocular surface neoplasms such as squamous cell carcinoma, basal cell carcinoma, and malignant melanoma may occur, with a predilection for the limbal area (Fig. 31.3B). Corneal changes include exposure keratitis, band-shaped nodular keratopathy, scarring, ulceration, vascularization, and perforation. The posterior segment is usually spared. Elevated symptomatic conjunctival nodules and suspected neoplasms may require repeated excisions; otherwise the treatment is symptomatic.
Sturge-Weber syndrome
This is a congenital disorder with a classical triad of cutaneous facial angioma, leptomeningeal angioma, and ocular involvement (see Chapter 65). The facial angioma typically occurs in the distribution of the ophthalmic division of the trigeminal nerve. Dilated episcleral and conjunctival vessels with aneurysm formation in the limbal area are commonly seen. Glaucoma is a frequent accompaniment (see Chapter 37), especially in patients with severe conjunctival involvement.3
Icthyosis
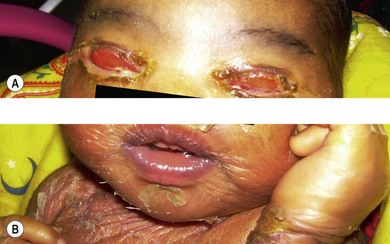
Ichthyosis is a heterogeneous family of at least 28 genetic skin disorders. Most pedigrees are either autosomal dominant or X-linked in their inheritance pattern. A rare autosomal recessive form, lamellar icthyosis, occurs. In all these conditions, dry scaly lesions are present predominantly over the upper half of the body, mainly around the neck, mouth, and trunk. The conjunctiva may become inflamed primarily or secondarily due to lid anomalies like ectropion.4 A papillary reaction may develop (Fig. 31.4). The treatment is to provide adequate lubrication and to correct the lid abnormalities, if present.
Alkaptonuria
This is a rare autosomal recessive disorder in which an affected individual’s urine turns a dark brown-blackish color when exposed to air. It is linked to chromosome 3q21-q24, caused by a deficiency of homogentisic 1,2-dioxygenase.5 This deficiency results in accumulation of homogentisic acid, which gets deposited in various tissues and organs. Systemic features include pigmentation over the face and nails, calcific and atherosclerotic heart disease, and arthritis. Ocular manifestations include a brown to black pigmentation of the nasal and temporal sclera especially in the area of the horizontal rectus muscle insertions. Pigmentation of the cornea has been reported.



