Chapter 51 Congenital optic disk anomalies
Introduction
A comprehensive evaluation of congenital anomalies of the optic disk needs an understanding of the ophthalmoscopic features, associated findings, pathogenesis, and ancillary studies for each anomaly.1 New ocular and systemic associations and theories of pathogenesis for many optic disk anomalies have emerged. Subclassification of excavated optic disk anomalies previously lumped together as colobomatous defects has further refined our ability to predict associated central nervous system (CNS) anomalies based on the appearance of the optic disk. High-resolution neuroimaging has refined prediction of subtle neurodevelopmental and endocrinologic associations of CNS anomalies.1
Four concepts are helpful in the management of congenital optic disk anomalies:
1. Children with bilateral optic disk anomalies present in infancy with poor vision and nystagmus: unilateral cases present during preschool years with sensory esotropia.
2. CNS malformations are common in patients with malformed optic disks.
3. Any structural ocular abnormality that reduces visual acuity in infancy may lead to amblyopia.2 A trial of occlusion is warranted in a child with asymmetric optic disk anomalies and decreased vision.
4. The finding of a discrete V- or tongue-shaped zone of infrapapillary retinochoroidal depigmentation with an anomalous optic disk should prompt a search for a trans-sphenoidal encephalocele.3
Optic nerve hypoplasia
Optic nerve hypoplasia is unquestionably the most common optic disk anomaly encountered in ophthalmologic practice in many countries.1 Many cases previously went unrecognized or were misconstrued as congenital optic atrophy. Parental drug and alcohol abuse, more widespread in recent years, contributes to an increasing prevalence of optic nerve hypoplasia.1,4 Teratogenic agents and systemic disorders associated with optic nerve hypoplasia are summarized in Box 51.1.
Box 51.1
Systemic and teratogenic associations with optic nerve hypoplasia
| Systemic associations | Risk factors |
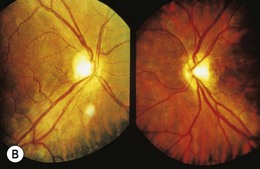
Ophthalmoscopically, optic nerve hypoplasia appears as an abnormally small optic nerve head, pink, gray or pale in color, and is often surrounded by a yellowish mottled peripapillary halo, bordered by a ring of increased or decreased pigmentation (the “double-ring” sign) (Fig. 51.1).4 The major retinal veins are often tortuous which may be helpful in establishing the diagnosis.5
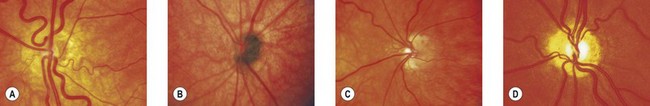
Histopathologically, optic nerve hypoplasia is a subnormal number of optic nerve axons with normal mesodermal elements and glial supporting tissue.6,7 The double-ring sign consists of the normal junction between the sclera and lamina cribrosa, corresponding to the outer ring, and an abnormal extension of retina and pigment epithelium over the outer portion of the lamina cribrosa, corresponding to the inner ring.6,7
Visual acuity in optic nerve hypoplasia ranges from logMAR 0.0 (6/6, 20/20, 1.0) to no light perception with localized visual field defects, often with a generalized constriction.8 Since visual acuity is determined primarily by the integrity of the papillomacular nerve fiber bundle, it does not necessarily correlate with the overall size of the disk. There is a strong association of astigmatism with optic nerve hypoplasia.9
Amblyopic eyes have smaller optic disks and smaller axial lengths compared to fellow eyes, suggesting that vision impairment in amblyopia may be caused by optic nerve hypoplasia with relative microphthalmos.10 This might be due to amblyopia being correlated with hyperopia and anisometropia11 but when axial length was measured, the optic disk areas of eyes with hyperopic strabismus with and without amblyopia were significantly reduced compared with hyperopic eyes without amblyopia or esotropia.12
Except when amblyopia develops in one eye, visual acuity usually remains stable throughout life. However, mild optic nerve hypoplasia occurs in children with congenital suprasellar tumors. A confusing diagnostic picture of acquired visual loss in the child with optic nerve hypoplasia may result.13
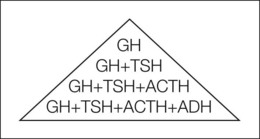
Optic nerve hypoplasia is often associated with CNS abnormalities. Septo-optic dysplasia (de Morsier’s syndrome) is the constellation of small anterior visual pathways, absence of the septum pellucidum, and thinning or agenesis of the corpus callosum;14 it may be associated with pituitary dwarfism.15 Isolated growth hormone, thyrotropin, corticotropin, or antidiuretic hormone deficiency may occur (Fig. 51.2).1,16 Hypothyroidism, panhypopituitarism, diabetes insipidus, and hyperprolactinemia may result.17–19 Growth hormone deficiency may be clinically inapparent in early life because high prolactin levels stimulate normal growth.20 Puberty may be precocious, or delayed, in children with hypopituitarism.21 Subclinical hypopituitarism can manifest as acute adrenal insufficiency following general anesthesia. It may be prudent to treat children who have optic nerve hypoplasia with perioperative corticosteroids.22
In an infant with optic nerve hypoplasia, a history of neonatal jaundice suggests congenital hypothyroidism, while neonatal hypoglycemia or seizures suggests congenital panhypopituitarism.4 Because of difficulties in measuring normal growth hormone levels that vary diurnally, most patients with optic nerve hypoplasia are followed clinically and investigated biochemically if growth is subnormal. However, when MRI shows posterior pituitary ectopia, or when a clinical history of neonatal jaundice or neonatal hypoglycemia is obtained, anterior pituitary hormone deficiency is probable, and more extensive endocrinologic testing becomes mandatory.23
Children with septo-optic dysplasia and corticotropin deficiency are at risk for sudden death during febrile illness,24 which may be caused by an impaired ability to increase corticotropin secretion in response to the stress of infection. They may have diabetes insipidus that contributes to dehydration during illness and hastens the development of shock. Some also have hypothalamic thermoregulatory disturbances signaled by hypothermia during well periods and high fevers during illnesses; they usually have had multiple hospital admissions for viral illnesses which can precipitate hypoglycemia, dehydration, hypotension, or fever of unknown origin.24 Because corticotropin deficiency represents the pre-eminent threat to life in children with septo-optic dysplasia, a complete anterior pituitary hormone evaluation, including provocative serum cortisol testing and assessment for diabetes insipidus, should be performed in children who have clinical symptoms (history of hypoglycemia, dehydration, or hypothermia) or neuroimaging signs (absent pituitary infundibulum with or without posterior pituitary ectopia) of pituitary hormone deficiency.
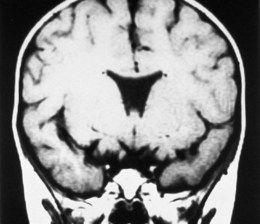
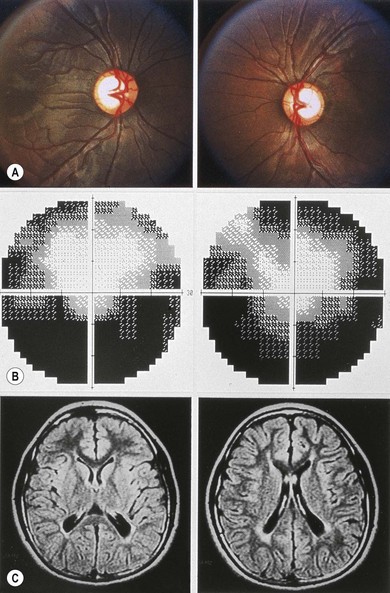
MRI delineates associated CNS malformations in patients with optic nerve hypoplasia.25 MRI provides high-contrast resolution and multiplanar imaging capability, allowing the anterior visual pathways to be visualized as distinct, well-defined structures.25 Coronal and sagittal T1-weighted MR images shows thinning and attenuation of the corresponding prechiasmatic intracranial optic nerve (Fig. 51.3). Coronal T1-weighted MRI shows diffuse thinning of the optic chiasm in bilateral optic nerve hypoplasia and focal thinning or absence of the side of the chiasm corresponding to the hypoplastic nerve in unilateral optic nerve hypoplasia.25 MRI may show a decrease in intracranial optic nerve size accompanied by other features of septo-optic dysplasia assisting in the diagnosis.25
Cerebral hemispheric abnormalities are evident in approximately 45% of patients with optic nerve hypoplasia (Fig. 51.4). They may consist of hemispheric migration anomalies (e.g. schizencephaly, cortical heterotopia), intrauterine or perinatal hemispheric injury (e.g. periventricular leukomalacia, encephalomalacia).26 Evidence of perinatal injury to the pituitary infundibulum (seen on MRI as posterior pituitary ectopia) is found in approximately 15% of patients with optic nerve hypoplasia.26 Normally, the posterior pituitary gland appears bright on T1-weighted images, because of the composition of the vesicles within it.23,26 In posterior pituitary ectopia, MRI demonstrates absence of the normal posterior pituitary bright spot and the pituitary infundibulum, and an ectopic posterior pituitary bright spot where the upper infundibulum is normally located (Fig. 51.5).23,26
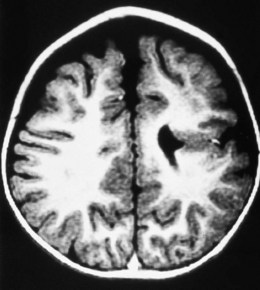
Fig. 51.4 Schizencephaly involving the left cerebral hemisphere.
(From Brodsky MC, Glasier CM. Optic nerve hypoplasia: clinical significance of associated central nervous system abnormalities on magnetic resonance imaging. Arch Ophthalmol 1993; 111: 66−74.)
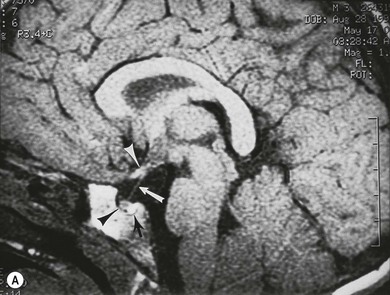
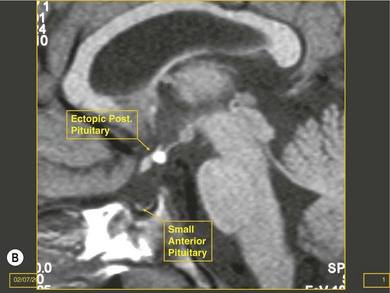
In optic nerve hypoplasia, posterior pituitary ectopia usually suggests anterior pituitary hormone deficiency; cerebral hemispheric abnormalities are predictive of neurodevelopmental deficits.26 Absence of the septum pellucidum does not portend neurodevelopmental deficits or pituitary hormone deficiency.27 Thinning or agenesis of the corpus callosum predicts neurodevelopmental problems by association with cerebral hemispheric abnormalities. The finding of unilateral optic nerve hypoplasia does not preclude coexistent intracranial malformations.26 Therefore, MRI is used to provide prognostic information in the child with optic nerve hypoplasia.26
Segmental optic nerve hypoplasia
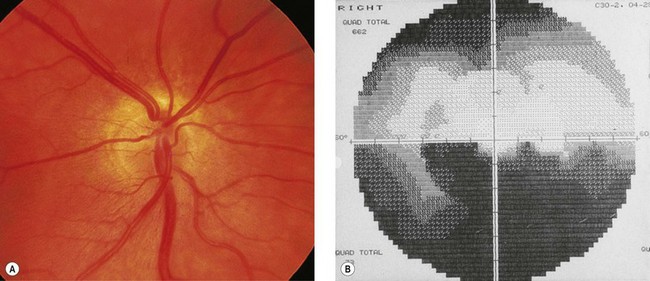
Some forms of optic nerve hypoplasia are segmental.28 “Superior segmental optic hypoplasia” (SSOH) with an inferior visual field defect occurs in some children of insulin-dependent diabetic mothers (Fig. 51.6).29,30 SSOH is usually an isolated anomaly31 and has an incidence of approximately 8%. The inferior visual field defects in superior segmental optic hypoplasia differ from typical nerve fiber bundle defects perhaps due to a regional impairment in retinal development.30 SSOH also occurs in patients whose mothers were not diabetic; it, therefore, is not pathognomonic for maternal diabetes.32 The mechanism by which insulin-dependent diabetes mellitus interferes with the early gestational development of superior retinal ganglion cells or their axons remains elusive.33 Mice lacking EphB receptor guidance proteins exhibit guidance defects in axons originating from the dorsal or superior part of the retina,34 which may explain this segmental hypoplasia.34
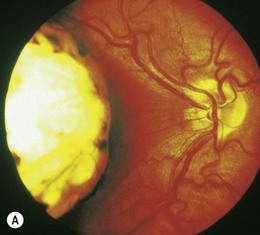
Congenital lesions of the retina, optic nerve, chiasm, tract, or retrogeniculate pathways are associated with segmental hypoplasia of the corresponding portions of the optic nerve (Fig. 51.7).28 Chiasmal hypoplasia produces focal loss of the nasal and temporal nerve fiber layer with hypoplasia of corresponding portions of the optic nerve (see Fig. 51.7). “Homonymous hemioptic hypoplasia” is an asymmetric form of segmental optic nerve hypoplasia seen in patients with unilateral congenital lesions of the postchiasmal afferent visual pathways.35 The nasal and temporal aspects of the optic disk contralateral to the hemispheric lesion show segmental hypoplasia and loss of the corresponding nerve fiber layer. There may be a central band of horizontal pallor across the disk. The ipsilateral optic disk may be normal in size or hypoplastic.28 Homonymous hemioptic hypoplasia results from trans-synaptic degeneration of the optic tract that is usually seen in congenital hemispheric lesions.28,35a Congenital suprasellar tumors can rarely produce a horizontal “bow-tie” cupping with selective loss of the nasal and temporal nerve fiber layer.35b
Periventricular leukomalacia and optic nerve hypoplasia
Periventricular leukomalacia (PVL) produces another form of optic nerve hypoplasia. PVL can be associated with abnormally large optic cups and a thin neuroretinal rim contained within normal-sized optic disks (Fig. 51.8).36 This may be due to intrauterine injury to the optic radiations with retrograde trans-synaptic degeneration of retinogeniculate axons after the scleral canals had established normal diameters. The large optic cups can simulate glaucoma, but the history of prematurity, normal intraocular pressure, and characteristic symmetric inferior visual field defects distinguish PVL from glaucoma.37 This may be a form of segmental optic nerve hypoplasia, although some believe it is a prenatal form of optic atrophy because of its normal optic disk diameter.37
The embryogenesis of optic nerve hypoplasia
At least two mechanisms may be operative in the embryogenesis of optic nerve hypoplasia:
1. A primary failure of retinal ganglion cell differentiation at the 13–15 mm stage of embryonic life (4–6 weeks of gestation).38
2. A deficiency of axon guidance molecules at the optic disk.
Netrin-1 is an axon guidance molecule expressed by neuroepithelial cells at the developing optic nerve head. Retinal ganglion cells in vitro respond to netrin-1 as a guidance molecule. Mice with a targeted deletion of the netrin-1 gene exhibit pathfinding errors at the optic disk. Retinal ganglion cells fail to exit into the optic nerve and optic nerve hypoplasia results.39–41 The lack of netrin-1 also results in abnormalities in other parts of the CNS (agenesis of the corpus callosum and axonal guidance defects in the hypothalamus).40 The timing of the CNS injuries suggests optic nerve hypoplasia results from intrauterine encephaloclastic destruction of a normally developed structure, whereas others represent a primary failure of axons to develop.16 In human fetuses, Provis et al. found a peak of 3.7 million optic axons at 16–17 weeks of gestation, with a subsequent decline to 1.1 million axons by the 31 weeks of gestation.42 This massive apoptotic loss of supernumerary axons may establish the correct functional topography of the visual pathways.4 Toxins or CNS injury could augment the processes by which superfluous axons are eliminated from the developing visual pathways.4,16,26 The association of optic nerve hypoplasia with periventricular leukomalacia26 cannot be reconciled with a deficiency of axon guidance molecules at the optic disk. It suggests retrograde trans-synaptic degeneration in the development of some forms of optic nerve hypoplasia.36,37
Cases of optic nerve hypoplasia in siblings43,44 are rare, and subsequent siblings of a child with optic nerve hypoplasia are at little additional risk. While genetic mutations in the human netrin-1 and DCC genes have not been described, homozygous mutations in the HESX1 gene have been identified in two siblings with optic nerve hypoplasia, absence of the corpus callosum, and hypoplasia of the pituitary gland.45 Five additional mutations in HESX1 have been observed in children with sporadic pituitary disease and septo-optic dysplasia46 which have clustered in the DNA-binding region of the protein consistent with a presumed loss in protein function. Examination of homeobox genes with expression patterns similar to Hesx1, such as Six3 and Six6, may yield additional genes responsible for both sporadic and familial septo-optic dysplasia.47 Optic nerve hypoplasia may accompany other ocular malformations in patients with mutations in the PAX6 gene.48
Excavated optic disk anomalies
Morning glory disk anomaly
In the morning glory disk anomaly and peripapillary staphyloma, an excavation of the posterior globe surrounds and incorporates the optic disk. In the other conditions, the excavation is contained within the optic disk. It is clear that optic disk colobomas, morning glory optic disks, and peripapillary staphylomas are distinct anomalies, each with a specific embryologic origin and not phenotypic spectrum variants.49
The morning glory disk anomaly is a congenital, funnel-shaped excavation of the posterior fundus that incorporates the optic disk49 which resembles a morning glory flower.50 The disk is markedly enlarged, orange or pink and may appear to be recessed or elevated centrally within a funnel-shaped peripapillary excavation (Fig. 51.9).49 A wide annulus of chorioretinal pigmentary disturbance surrounds the disk49 and a white glial tuft overlies its central portion. The blood vessels appear increased in number and arise from the periphery of the disk,49 curving abruptly as they leave the disk to run an abnormally straight course over the peripapillary retina. It is often difficult to distinguish arterioles from venules. Small peripapillary arteriovenous communications may occur.51 The macula may be incorporated into the excavation (“macular capture”).52 Neuroimaging shows a funnel-shaped enlargement of the distal optic nerve at its junction with the globe.1
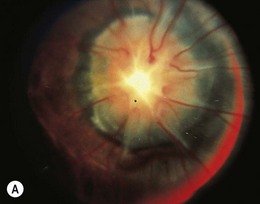
The morning glory disk anomaly is usually unilateral, but several bilateral cases have been reported.49,52 Visual acuity usually ranges from logMAR 1.0 (6/60, 20/200, 0.1) to “finger counting,” although cases with 20/20 vision as well as no light perception have been reported. Amblyopia may contribute to visual loss2 and a trial of occlusion therapy is warranted in children. Optic disk colobomas have no racial or gender predilection: morning glory disks are more common in females and rare in black people.
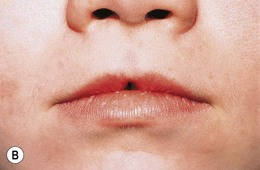
The morning glory disk anomaly is associated with a transsphenoidal basal encephalocele.52–54 It may be accompanied by a V- or tongue-shaped zone of infrapapillary depigmentation, a sign suggesting a trans-sphenoidal encephalocele (see Fig. 51.9).3 Trans-sphenoidal encephalocele is a rare midline congenital malformation in which a meningeal pouch, often containing the chiasm and adjacent hypothalamus, protrudes through a defect in the sphenoid bone (Fig. 51.10). Children with this basal meningocele have a wide head, a flat nose, mild hypertelorism, a midline notch in the upper lip, and sometimes a midline cleft in the soft palate (Fig. 51.11). The meningocele protrudes into the nasopharynx and may obstruct the airway. Symptoms including rhinorrhea, nasal obstruction, mouth breathing, or snoring55,56 may be overlooked unless the morning glory disk anomaly or the facial configuration is recognized. A trans-sphenoidal encephalocele may appear as a pulsatile posterior nasal mass or as a “nasal polyp” high in the nose. Surgical biopsy or excision can be lethal.55 Associated brain malformations include agenesis of the corpus callosum and posterior dilatation of the lateral ventricles. Absence of the chiasm (achiasmia) is seen in approximately one-third of patients. Most of the affected children have no overt intellectual or neurologic deficits, but panhypopituitarism is common.55 Surgery for trans-sphenoidal encephalocele may be contraindicated, since herniated brain tissue can include vital structures.1,3
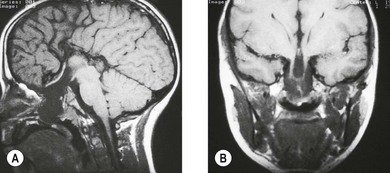

The morning glory disk anomaly can be associated with hypoplasia of the ipsilateral intracranial vasculature.57–59 With MR angiography, ipsilateral intracranial vascular dysgenesis (with or without Moyamoya syndrome) is seen in some patients with morning glory disk anomaly (Fig. 51.12).57–59 This suggests that the morning glory disk anomaly results from a primary vascular dysgenesis as part of a regional mesodermal dysgenesis.60
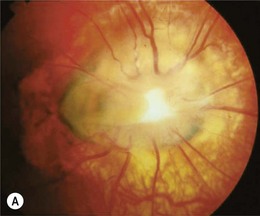
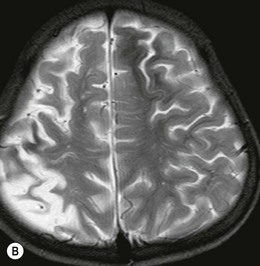
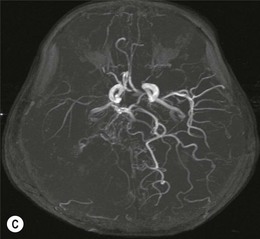
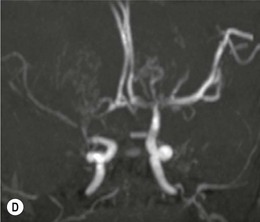
Figure 51.12 Moyamoya disease in a child with a monocular morning glory disk anomaly. Reduced perfusion of the right hemisphere.
(Neuroimages courtesy of Dr Roxana Gunny. MGDA photograph from another patient.)
Usually, the morning glory disk anomaly is not part of a genetic disorder1,50 but it has been associated with ipsilateral orofacial hemangioma.61 This association may fall within the spectrum of the PHACE syndrome (posterior fossa malformations, large facial hemangiomas, arterial anomalies, cardiac anomalies and aortic coarctation, and eye anomalies) which occurs only in girls.62 Associated ipsilateral intracranial vascular dysgenesis (Fig. 51.13) supports this.63 Atypical morning glory disk anomalies have also rarely been reported in patients with neurofibromatosis 2.64
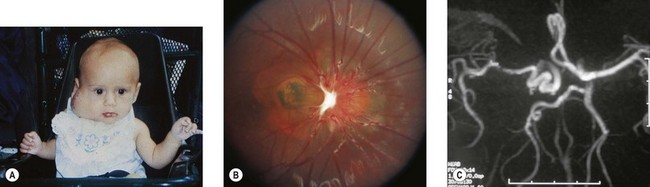
Serous retinal detachments develop in 26–38% of eyes with morning glory optic disk anomalies;49 they originate in the peripapillary area and extend through the posterior pole, occasionally progressing to total detachments. Small retinal tears adjacent to the optic nerve in patients with morning glory disk-associated retinal detachments have been identified,65 and subretinal neovascularization may develop adjacent to a morning glory disk.66 Contractile movements occur in morning glory optic disks,49 perhaps due to fluctuations in subretinal fluid volume altering the degree of retinal separation within the excavation.49 One patient had episodes of amaurosis with transient dilation of the retinal veins in an eye with a morning glory disk.67
The embryologic defect leading to the morning glory disk anomaly is widely disputed.68 Unlike optic disk coloboma (discussed below), it does not result from defective closure of the embryonic fissure.68–70 The central glial tuft, vascular anomalies, and a scleral defect, together with the histologic findings of adipose tissue and smooth muscle within the peripapillary sclera perhaps signify a primary mesenchymal abnormality. The midfacial anomalies in some patients support a primary mesenchymal defect: most of the cranial structures are derived from mesenchyme.68 The basic defect may be mesodermal, but some features may result from a dynamic disturbance between the relative growth of mesoderm and ectoderm.61
The symmetry of the fundus excavation with respect to the disk implicates an anomalous funnel-shaped enlargement of the distal optic stalk at its junction with the primitive optic vesicle, as the primary embryologic defect.49 This hypothesis suggests that the glial and vascular abnormalities that characterize the morning glory disk anomaly are secondary effects of a primary neuroectodermal dysgenesis on the formation of mesodermal elements that arise later in embryogenesis.49
Optic disk coloboma
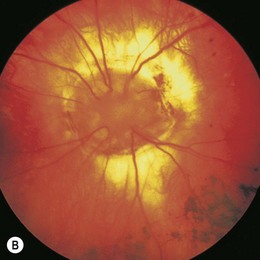
The term coloboma (koloboma in Greek) means curtailed or mutilated.71 In optic disk coloboma, a sharply delimited, glistening white, bowl-shaped excavation occupies an enlarged optic disk (Fig. 51.14). The excavation is decentered inferiorly, reflecting the position of the fetal fissure relative to the primitive epithelial papilla.49 The inferior neuroretinal rim is thin or absent while the superior neuroretinal rim is relatively spared. Rarely, the entire disk may appear excavated; however, the colobomatous nature of the defect can still be appreciated since the excavation is deeper inferiorly.49 The defect may extend further inferiorly to involve the adjacent choroid and retina, in which case microphthalmia is frequently present.72 Iris and ciliary colobomas often coexist. Axial scanning shows a crater-like excavation of the posterior globe at its junction with the optic nerve.69
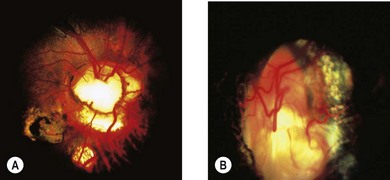
Visual acuity may be decreased and is difficult to predict from the appearance of the disk.1 Unlike the morning glory disk anomaly, which is usually unilateral, optic disk colobomas occur equally unilaterally or bilaterally.49 Optic disk colobomas may arise sporadically or be inherited in an autosomal dominant fashion. They may be accompanied by multiple systemic abnormalities in a myriad of conditions including the CHARGE association,73 Walker-Warburg syndrome, Goltz’ focal dermal hypoplasia, Aicardi’s syndrome, Goldenhar’s syndrome, and linear sebaceous nevus syndrome.1 Rarely, large orbital cysts occur in conjunction with atypical excavations of the disk. These are probably colobomatous.72,74 The cyst may communicate with the excavation.74,75 Intrascleral smooth muscle strands oriented concentrically around the distal optic nerve76 may account for the contractility of the optic disk seen rarely in optic disk colobomas.77
Eyes with isolated optic disk colobomas can develop serous macular detachments in contrast to the rhegmatogenous retinal detachments that complicate retinochoroidal colobomas,78,79 perhaps from diffusion of retrobulbar fluid into the subretinal space.79 Treatment includes patches, bedrest, corticosteroids, vitrectomy, scleral buckling procedures, gas-fluid exchange, and photocoagulation.80,81 Spontaneous reattachment may occur.81
Coronal T1-weighted MRI confirms that the intracranial portion of the optic nerve is reduced in size.81 Many uncategorizable dysplastic optic disks are indiscriminately labeled as optic disk colobomas.82 This complicates the nosology of coloboma-associated genetic disorders. It is crucial that the diagnosis of optic disk coloboma be reserved for disks that show an inferiorly decentered, white-colored excavation with minimal peripapillary pigmentary changes.1,49 In striking contrast to the numerous well-documented reports of morning glory optic disks occurring in conjunction with basal encephaloceles, cases of optic disk coloboma with basal encephalocele are conspicuous by their absence.83,84
Although the phenotypic profiles of optic disk coloboma and the morning glory disk anomaly may overlap, the ophthalmoscopic features of optic disk coloboma (Table 51.1) are most consistent with a primary structural dysgenesis involving the proximal fetal fissure, as opposed to an anomalous dilatation confined to the distal optic stalk in the morning glory disk anomaly.49 The differences in associated ocular and systemic findings between the two anomalies (Table 51.2) support this hypothesis.1 Anomalous optic disks with features of both are occasionally seen: they may represent early embryonic injury involving both the proximal fetal fissure and the distal optic stalk. However, the great majority of colobomatous and morning glory optic disks are distinct.
Table 51.1 Ophthalmoscopic findings that distinguish the morning glory disk anomaly from optic disk coloboma
| Morning glory disk | Optic disk coloboma |
|---|---|
| Optic disk lies within the excavation | Excavation lies within the optic disk |
| Symmetrical defect (disk lies centrally within the excavation) | Asymmetrical defect (excavation lies inferiorly within the disk) |
| Central glial tuft | No central glial tuft |
| Severe peripapillary | Minimal peripapillary |
| Pigmentary disturbance | Pigmentary disturbance |
| Anomalous retinal vasculature | Normal retinal vasculature |
Table 51.2 Associated ocular and systemic findings that distinguish the morning glory disk from isolated optic disk coloboma
| Morning glory disk | Optic disk coloboma |
|---|---|
| More common in females; rare in Blacks | No sex or racial preference |
| Rarely familial | Often familial |
| Rarely bilateral | Often bilateral |
| No iris, ciliary, or retinal colobomas | Iris, ciliary, and retinal colobomas common |
| Rarely associated with multisystem genetic disorders | Often associated with multisystem genetic disorders |
| Basal encephalocele common | Basal encephalocele rare |
Colobomas are most often sporadic but may be autosomal dominant, autosomal recessive, or X-linked recessive. One study found a 10% recurrence rate, but the true percentage of colobomas that are inherited is probably higher.85 A wide variety of mutations have been documented in patients with colobomas. Except for the CHD7 mutation, which accounts for 60% of cases of CHARGE syndrome,86 no mutations predominate and no “genetic panel” exists. For this reason, genetic testing is probably unwarranted unless there are other dysmorphic features.
Peripapillary staphyloma
Peripapillary staphyloma is an extremely rare, usually unilateral, anomaly in which a deep fundus excavation surrounds the optic disk.87,88 The disk is at the bottom of the excavated defect and may appear normal or show temporal pallor (Fig. 51.15).88 The walls and margin of the defect may show atrophic pigmentary changes involving the RPE and choroid.88 There is no central glial tuft overlying the disk and the retinal vascular pattern is normal. The staphylomatous excavation is deeper than that in the morning glory disk anomaly. Several cases of contractile peripapillary staphyloma have been documented,89–91 sometimes with transient visual obscurations.92
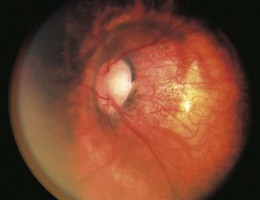
Fig. 51.15 Peripapillary staphyloma.
(From Brodsky MC. Congenital optic disk anomalies. Surv Ophthalmol 1994: 89−112.)
Visual acuity is usually reduced, but normal acuity has been reported.93 Affected eyes are usually emmetropic or slightly myopic.87 Eyes with decreased vision frequently have centrocecal scotomas.87 Although peripapillary staphyloma is usually unassociated with systemic or intracranial disease, it has been reported in association with trans-sphenoidal encephalocele,94 PHACE syndrome,62,95 linear nevus sebaceous syndrome, and 18q- syndrome.96
The relatively normal appearance of the optic disk and retinal vessels in peripapillary staphyloma suggests that the development of these structures is complete prior to the onset of the staphylomatous process.49 The clinical features of peripapillary staphyloma are consistent with diminished peripapillary structural support, perhaps resulting from incomplete differentiation of sclera from posterior neural crest cells. Staphyloma formation presumably occurs when establishment of normal intraocular pressure leads to herniation of unsupported ocular tissues through the defect.49 Peripapillary staphyloma and the morning glory disk anomaly appear to be pathogenetically distinct both in the timing of the insult (5 months’ gestation vs. 4 weeks’ gestation) as well as the embryologic site of structural dysgenesis (posterior sclera versus distal optic stalk).



