Nasal Embryology
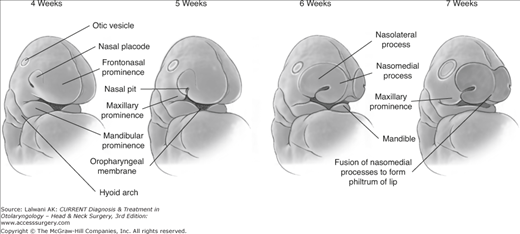
Nasal development occurs during weeks 4 through 10 of gestation. Migrating neural crest cells populate the frontonasal prominence, one of five facial prominences, and form the nasal or olfactory placodes. These placodes appear as convex thickenings on the surface ectoderm of the frontonasal prominence. A central depression deepens in the placodes to form the primitive nasal pit. Mesenchymal proliferation during the 5th week around the nasal placodes allows the horseshoe-shaped medial and lateral nasal prominences to develop and fuse to form the nostrils. The nasal pits grow toward the oral cavity and develop into the early nasal fossae. The nasobuccal membrane separates the nasal cavities from the oral cavity. This membrane subsequently disappears, allowing for communication of the nasal cavities with the oral cavity, forming the primitive posterior nasal choanae. The nasomedial process gives rise to part of the nasal septum and the medial crus of the lower lateral alar cartilage. The nasolateral process develops into the external wall of the nose, nasal bones, upper lateral cartilage, alae, and lateral crus of the lower lateral cartilage. The apex and dorsum of the nose develop from the frontonasal process (Figure 11–1).
The proposed classification of congenital nasal deformities separates them into four categories. Type I deformities represent hypoplasia and atrophy, Type II are hyperplasia and duplications, Type III are clefts, and Type IV deformities consist of neoplasms and vascular anomalies.
Arrhinia
Arrhinia is a rare congenital absence of the nose. The findings include absence of nasal bones, cribriform plate, and nasal septum. In cases of total arrhinia, the olfactory system is also absent. Arrhinia can be associated with other craniofacial anomalies and midline defects.
The likely embryologic abnormality resulting in arrhinia is thought to be a failure of the nasal placodes to invaginate during the 5th week of fetal development. Most cases reported are sporadic, although cases of genetic aberration have been described.
A computed tomography (CT) or magnetic resonance imaging (MRI) scan is performed to plan for surgery and often reveals associated abnormalities. Radiological examination demonstrates small or absent nasal bones and bony masses obstructing the nasal cavity.
All cases require airway management in the neonatal period. Reconstructive surgery is usually postponed until 4–6 years of age. Surgery is often performed in multiple stages, using techniques such as forehead flaps, rib grafts, and tissue expansion.
Heminose
Unilateral nostril agenesis is a rare malformation frequently associated with other facial anomalies, including proboscis lateralis, abnormalities of the lacrimal system, and malformations of facial bones. Etiology of heminose is unclear, and in the reported cases, this malformation has occurred sporadically. The lack of a nasal placode is thought to lead to heminasal abnormality. Radiographic examination may reveal unilateral absence of the cribriform plate. Surgical reconstruction is usually performed around 4–6 years of age and involves a complex multistaged procedure.
Nasal Pyriform Aperture Stenosis
- Presents with respiratory difficulty due to nasal obstruction at birth or shortly thereafter.
- Examination reveals bony obstruction at the nasal vestibule.
- CT scan is usually performed to confirm the diagnosis.
Congenital nasal pyriform aperture stenosis is a rare cause of airway obstruction in newborns and was first described in 1989. A bony overgrowth of the medial maxilla leads to narrowing of the nasal inlet. Because neonates are obligate nasal breathers, presenting signs and symptoms include respiratory distress, cyclical cyanosis or apnea that is relieved with crying, feeding difficulties, and, in severe cases, life-threatening total airway obstruction. Depending on severity of stenosis, symptoms can occur at birth or shortly thereafter. Examination of the nose reveals a bony obstruction in the vestibule and an inability to pass a catheter or endoscope into the nose. Pyriform aperture stenosis can be found either in isolation or together with other malformations, including submucous cleft palate, absence of the anterior pituitary gland, hypoplastic maxillary sinuses, hypotelorism, and a flat nasal. Up to 60% of nasal pyriform aperture stenosis is associated with a single central maxillary incisor. These patients should be further evaluated for holoprosencephaly.
A CT scan through the midface is usually performed to confirm the diagnosis and delineate the anomaly. On CT imaging, nasal pyriform aperture stenosis is diagnosed when the transverse diameter of each aperture is less than 3 mm or combined aperture width is less than 8 mm. In addition, brain MRI or CT allows evaluation of associated pituitary or midbrain abnormalities.
Management of congenital nasal pyriform aperture stenosis depends on the severity of the symptoms. The primary goal is to establish a safe airway. In mild cases, nasal obstruction can be managed conservatively with topical nasal decongestants, corticosteroids, suctioning, or humidification. Symptoms may resolve as the child continues to grow. In severe cases, a McGovern nipple or oral airway can be used. If the infant fails to respond to medical treatment, loses weight, has cyclical cyanosis, or develops pulmonary hypertension from the obstruction, surgical repair is recommended. Transnasal and sublabial approaches to pyriform aperture stenosis repair have been described, sublabial being a preferred method.
Choanal Atresia
- Bilateral cases present at birth with respiratory distress.
- Unilateral cases may present later in life with unilateral nasal obstruction or nasal discharge.
- CT scan confirms the diagnosis.
Choanal atresia is a congenital obstruction of the posterior nasal apertures. This abnormality occurs in one out of every 5000–7000 live births and affects females twice as often as males. It can occur unilaterally or bilaterally with unilateral atresia being more common. Choanal atresia has been described as bony, membranous, or mixed membranous-bony, with the mixed membranous–bony atresias being most common and occurring in 70% of cases. Possible etiology of choanal atresia includes persistence of the buccopharyngeal membrane from the foregut, failure of perforation of the nasobuccal membrane, abnormal persistence or location of mesoderm forming adhesions in the nasochoanal region, and misdirection of neural crest cell migration.
Choanal atresia is associated with other anomalies in half of the cases. The most commonly described association is with CHARGE (Coloboma of the eye, Heart malformations, Choanal Atresia, Retarded growth or development, Genital or urinary abnormalities, Ear malformations or deafness). Increased rates of choanal atresia have been associated with abnormalities in vitamin A metabolism and with prenatal use of thionamides (eg, methimizole or carbimizole).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


