Chapter 82 Congenital cranial dysinnervation disorders
The congenital cranial dysinnervation disorders (CCDDs) result from abnormal development of individual or multiple cranial nerve nuclei or their axonal connections. Most are either proven or suspected to have a genetic basis.1,2 Although many of these conditions are characterized only by abnormal extraocular muscle innervation, facial innervation, or both, others have associated systemic and/or neurologic abnormalities, such as cerebrovascular, cardiovascular, and skeletal malformations that also are attributable to the underlying genetic defect.2 At present, a total of seven disease genes and ten phenotypes have been identified.2 In this chapter, only those CCDDs characterized wholly or in part by abnormalities of eye movement will be discussed. These include the three main types of congenital fibrosis of the extraocular muscles (CFEOM), Duane’s retraction syndromes (DRS), homeobox A1 (HOXA1) spectrum, horizontal gaze palsy with progressive scoliosis (HGPPS), and Möbius’ syndrome.
Congenital fibrosis of extraocular muscles
This group of disorders is characterized by congenital ocular motility abnormalities, congenital ptosis, and fibrotic extraocular muscles. To date, three primary CFEOM main phenotypes have been described − CFEOM1, CFEOM2, and CFEOM3 − with CFEOM3 having three phenotypic variants: CFEOM3A, CFEOM3B, and CFEOM3C.2,3
Congenital fibrosis of the extraocular muscles type 1 (MIM #135700)
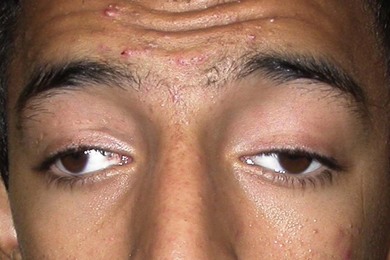
CFEOM1 is the most common of the CFEOM phenotypes. It is autosomal dominant with full penetrance and has been reported worldwide.4 It is characterized by congenital bilateral ptosis and a bilateral ocular motility disorder with the globes infraducted in primary position, restricted upgaze, and variably restricted horizontal gaze. These abnormalities result in many affected individuals adopting a chin-up head posture (Fig. 82.1). These patients have positive forced ductions and misdirected eye movements such as a marked synergistic convergence on attempted upgaze. They are rarely noted to have retraction. Neuropathologic studies reveal significant atrophy and fibrosis of the superior rectus and levator palpebrae muscles, with variable reduction in the size of the other extraocular muscles. The ocular motor nerves are absent or hypoplastic, and the diameter of the optic nerves is reduced by 30−40%.5 CFEOM1 is caused by a heterozygous missense mutation in KIF21A, a gene located on chromosome 12 (12q12).3 This gene encodes a kinesin microtubule-associated protein associated with anterograde organelle transport in neurons.6

Congenital fibrosis of the exraocular muscles type 2 (MIM #602078)
CFEOM2 is an autosomal recessive disorder characterized by bilateral ptosis associated with bilaterally absent adduction, upgaze, and downgaze (Fig. 82.2).7 Although abduction is present, it is limited and the pupils are often irregularly shaped and sized and non-reactive to both light and near stimulation. The appearance is that of bilateral oculomotor (third) nerve palsies and neuroimaging has shown what appears to be bilateral absence of the third nerves.8 This condition is caused by homozygous mutations in the PHOX2A gene located on chromosome 11 (11q13.3−q13.4)7 that codes for a homeodomain transcription factor expressed predominantly in developing oculomotor and trochlear neurons and that is essential for their survival. Mutations alter the factor sufficiently that the oculomotor (and probably trochlear) nerves never develop.
Congenital fibrosis of the extraocular muscles type 3
CFEOM3 is an autosomal dominantly inherited disorder that has clinical features similar to those of CFEOM1 except that they are more variable and sometimes associated with the ability to elevate the eyes above the midline.9 The phenotypic variability results, at least in part, because the condition may be caused by heterozygous mutations in at least two genes: TUBB3 (CFEOM3A; MIM #600638) and, rarely, KIF21A (CFEOM3B).10,11
Several different missense mutations may occur in the TUBB3 gene which codes for a component of microtubules. Thus, some patients with CFEOM3A have isolated ocular motor deficits (some even have isolated absence of upgaze), whereas others have bilateral facial weakness, peripheral sensory or sensorimotor neuropathies, wrist and finger contractures, cognitive dysfunction, or a combination of these features. Magnetic resonance imaging has shown dysgenesis of the corpus callosum and anterior commissure in some cases.11 Patients have been described who have a phenotype similar to CFEOM1 except for the presence of some upgaze and who have a mutation in KIF21A. They are said to have CFEOM3B.2 A single family has been described in which three generations have what appears to be CFEOM3 but who carry a reciprocal translocation involving chromosomes 2q and 13q,12 termed CFEOM3C (MIM #609384).
Practical management of CFEOM patients
Once the diagnosis of CFEOM is made, it is crucial to help the patients and their parents live with their complex and unremitting problems, particularly when they have not only ocular motor deficits and ptosis, but also systemic and/or neurologic deficits. The outlook is guarded, and, as the condition is rare, even the most experienced pediatric ophthalmologist can offer little in the way of a confident individual prognosis. Nevertheless, all children with CFEOM should undergo a refraction, be treated for amblyopia with part-time occlusion therapy or atropinization when appropriate (see Chapter 70), and be evaluated periodically throughout their period of visual development. In patients whose ptosis requires surgical treatment, one must be careful not to overcorrect, as this may result in exposure keratopathy because of the restricted eye movements and lack of a Bell’s phenomenon in most of these individuals. We find that a sling procedure is usually more effective than a levator resection and, in addition, allows overcorrection to be more easily managed. If a decision is made to perform a levator resection, the eyelids should be raised only so that they are above the pupillary axis.
Unfortunately, the strabismus associated with CFEOM is not particularly amenable to surgery. Results are poor because of the marked lack of ocular motility even when the eyes are able to be aligned in primary position by performing very large recessions and occasionally resections.13 Thus, the patient’s parents should be counseled accordingly so that they have realistic expectations regarding the goals of the surgery.
Duane’s retraction syndrome
Duane’s retraction syndrome (DRS) is the most common CCDD affecting ocular motility.2 There are several variants; however, the most common presentation is characterized by marked limitation or absence of abduction, variable limitation of adduction, and palpebral fissure narrowing with globe retraction on attempted adduction (Figs 82.3–82.5). Vertical ocular movements are often noted on adduction, most frequently in an upward direction (see Fig. 82.5).

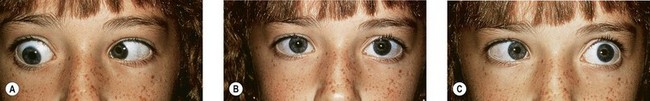
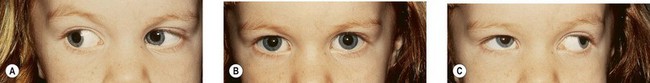
In most cases, DRS is unilateral (see Figs 82.3 and 82.5), but bilateral DRS occurs in 15–20% of affected patients (see Fig. 82.4). The syndrome occurs more commonly in females than males, and the left eye is more frequently affected than the right. Gaze is usually directed toward the side of the unaffected eye and, in some instances, the face is turned toward the affected side to allow binocular single vision. Visual symptoms are conspicuous by their absence. Vision is almost always normal unless there is associated anisometropia and amblyopia. Thus, in the majority of cases, no treatment is necessary unless the patient has a marked head turn (Fig. 82.6).
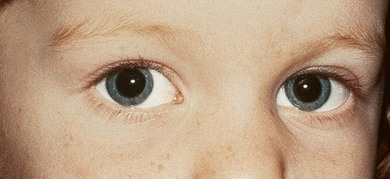
Fig. 82.6 Unilateral (right) Duane’s retraction syndrome type 2. Same patient as that in Fig. 82.5 showing patient’s typical head position consisting of a face turn to the left to maintain binocularity.
Early histologic studies demonstrated abnormalities of the lateral or medial rectus muscles and led investigators to conclude that DRS was a local, myogenic phenomenon. It was generally believed that the cause of the abduction deficiency was fibrosis of the lateral rectus muscle and that limitation of adduction was caused by a posterior insertion of the medial rectus muscle. Adhesions between the medial rectus muscle and the medial orbital wall were also reported. Subsequently, electromyographical studies, magnetic resonance imaging, and histological studies established that the disorder is caused by absence or hypoplasia of the abducens nerve with innervation of the ipsilateral lateral rectus muscle by a branch of the oculomotor nerve.14–16
A modest number of patients with DRS have associated congenital neurologic deficits that localize to the brainstem, such as crocodile tears and sensorineural hearing loss or structural defects involving ocular, skeletal, and neural structures.2
Most cases of DRS are sporadic, but familial unilateral and bilateral cases occur.17 Familial DRS usually is transmitted in an autosomal dominant pattern. Affected individuals in such families show substantial clinical diversity.
Duane’s retraction syndrome 1 (MIM #126800)
In the DURS1 phenotype, the syndrome may be unilateral (see Fig. 82.3) or bilateral (see Fig. 82.4) and, depending on the size of the deletion, may occur in association with other features including mental retardation, branchio-oto-renal syndrome, genital tract anomalies, and other somatic mutations. The gene disrupted by overlapping cytogenic abnormalities at the DURS1 locus located on chromosome 8(8q13) may be CPAH, a carboxypeptidase gene with eight exons.18,19
Duane’s retraction syndrome 2 (MIM #604356)
In the DURS2 phenotype, the condition is almost always bilateral (Fig. 82.5). Associated findings can include a variety of vertical deviations such as apparent underaction of the superior oblique and dissociated vertical deviation. Patients have been noted to have decreased abduction with or without decreased adduction. No patients have had adduction defects without abduction defects. Amblyopia is common. No associated somatic abnormalities have been found. Inheritance is autosomal dominant. The responsible gene, CHN1, is located on chromosome 2 (2q31).20–22 It is involved with ocular motor path finding during the development of the abducens nerve and, to a lesser extent, the oculomotor nerve.23,24



