Normal anatomy, embryology, and congenital anomalies of the esophagus are discussed in this article. The classification, epidemiology, embryology, diagnosis, and management, including outcome following repair of esophageal atresia with or without an associated tracheoesophageal fistula, are described. The diagnosis and management of less common anomalies, such as congenital esophageal stenosis and congenital esophageal duplication, are outlined.
Normal anatomy and embryology of the esophagus
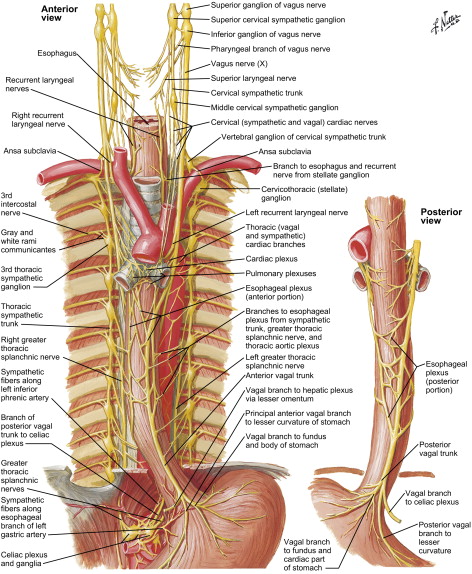
The esophagus is a hollow muscular tube consisting of mucosa, submucosa, and muscularis layers. The mucosa is composed of stratified squamous nonkeratinized epithelium that transitions to columnar epithelium at the gastroesophageal junction. The esophagus lacks a serosal layer. The muscle layer consists of striated muscle in the upper one third and smooth muscle in the lower two thirds of the tube; these are arranged as an inner circular and an outer longitudinal layer ( Fig. 1 ). During ingestion of a meal, distention of the proximal striated muscle initiates peristalsis, which continues down the esophagus and is coordinated by the medullary centers (nucleus ambiguus and dorsal motor nucleus) and the vagus nerve ( Fig. 2 ). The Auerbach’s (myenteric) plexus is found between the circular and longitudinal muscles and mediates intrinsic motor control of the esophagus, whereas the Meissner’s plexus is located within the submucosa and innervates the lamina muscularis mucosae; these are part of the parasympathetic nervous system.
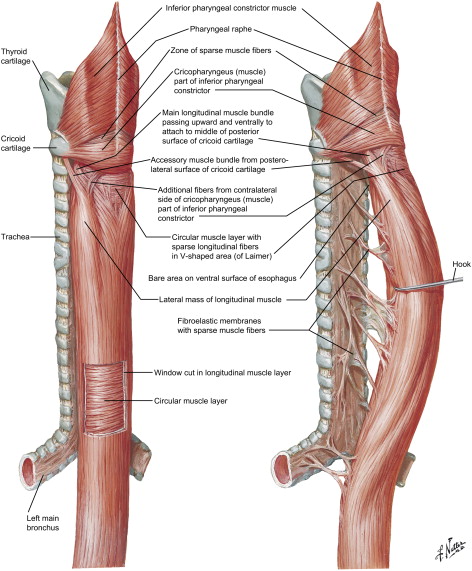
Two sphincters control passage of contents into the gastrointestinal tract: an anatomic upper esophageal sphincter (UES) and a physiologic lower esophageal sphincter (LES). The UES consists of cricopharyngeus and inferior pharyngeal constrictors, whereas the LES is histologically indistinguishable from the smooth muscle fibers of the lower esophagus.
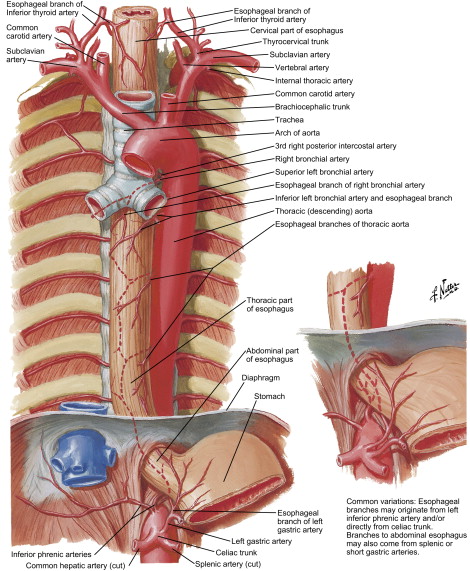
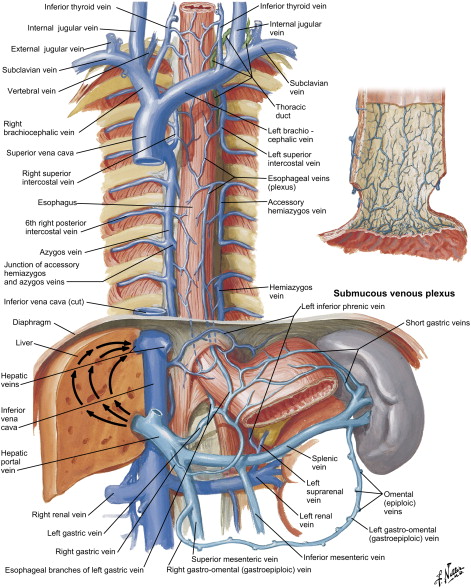
Blood supply of the esophagus is segmental and is dictated by its anatomic location: the cervical portion receives blood supply from branches of the inferior thyroid artery, the thoracic portion from bronchial arteries and direct branches from the aorta, and the abdominal portion from the left gastric and the inferior phrenic arteries ( Fig. 3 ). Venous drainage from the submucosal venous plexus follows the arterial supply in the cervical and abdominal portions, whereas the thoracic esophagus drains into the azygous and hemiazygous system ( Fig. 4 ). Likewise, the lymphatic drainage of the esophagus is segmental, with the cervical portion draining into deep cervical lymph nodes, the thoracic portion into the superior and posterior mediastinal lymph nodes, and the abdominal portion into the gastric and celiac lymph nodes.
The embryogenesis of esophageal atresia (EA) and tracheoesophageal fistulae is not completely understood. During fetal development, the foregut is formed around the 20th day of gestation. On day 22, the pharyngeal groove appears in the ventral aspect of the primitive foregut and within the next few days the lung bud forms at this locus. The mesenchyme between the foregut and the developing bronchial tree gives rise to the tracheoesophageal septum, and its proper development is believed to be necessary for the normal separation of the digestive and respiratory systems ( Fig. 5 ).
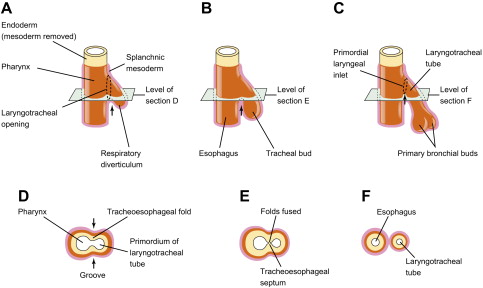
The classic theory of division of respiratory and digestive tracts proposes that the tracheoesophageal septum forms as a pair of lateral folds that fuse in the midline to divide the esophagus from the trachea; this theory assumes that the lung buds develop from the trachea. More recent theories propose the development of paired symmetric diverticula from the primitive foregut in the absence of evidence for lateral folds forming a tracheoesophageal septum .
Esophageal atresia and tracheoesophageal fistula
History
EA was first reported by William Durston in 1670, and in 1697 Thomas Gibson described EA with tracheoesophageal fistula (TEF). The management of such malformations proved to be challenging, with the first unsuccessful attempt at TEF ligation in 1913 by Harry Richter who performed a transpleural ligation of the fistula and a gastrostomy to provide nutrition. From 1936 to 1938 Thomas Lanman and Robert Shaw independently attempted extrapleural ligations of TEF with primary anastomoses in five separate cases, all resulting in eventual death in the immediate postoperative period. The first survivors were reported independently by N. Logan Leven and William Ladd in 1939 using a multistaged approach that included placement of a gastrostomy tube, extrapleural division of TEF, and cervical esophagostomy. Gastrointestinal continuity was established using an antethoracic skin tube from the gastrostomy to esophagostomy. Although continuity was established, the repaired esophagus did not have normal peristalsis .
In 1941 Cameron Haight performed the first successful primary repair of EA with TEF after initial resuscitation of his 12-day-old patient by ligating the TEF using a left extrapleural approach followed by end-to-end single layer anastomosis with interrupted silk sutures . The operative treatment today still uses concepts used by these pioneering surgeons, with the biggest advancements being made in the supportive management of the postoperative neonate. The rates of survival have significantly improved from approximately 50% in the 1940s to greater than 90% today .
Classification
Initial classification system for EA was developed by E.C. Vogt in 1929. This system was slightly modified and the numerical nomenclature was transformed into an alphabetic one by Gross. This classification system groups the anomalies into: EA without TEF (type A), EA with proximal TEF (type B), EA with distal TEF (type C), EA with TEF between both esophageal segments and trachea (type D), TEF without EA or H-type fistula (type E), and esophageal stenosis (type F) ( Fig. 6 ). Another classification system developed by Waterston and colleagues in 1962 focused on risk factors and predicted survival in infants who have EA ( Table 1 ). In the 1990s, Spitz and colleagues included birth weight and presence of cardiac anomalies and proposed a system that is currently used as an alternative to the Waterston classification ( Table 2 ).
| Group | Birth weight | General health | Survival |
|---|---|---|---|
| A | >2500 g | Otherwise healthy | 100% |
| B | 2000–2500 g | Otherwise healthy | 85% |
| >2500 g | Moderate associated anomalies (noncardiac, PDA, VSD, ASD) | ||
| C | <2000 g | Otherwise healthy | 65% |
| >2000 g | Severe associated cardiac anomalies |
| Group | Characteristics | Survival rate |
|---|---|---|
| I | Birth wt >1500 g, no major cardiac defects | 97% |
| II | Birth wt <1500 g or major cardiac defects | 59% |
| III | Birth wt <1500 g, major cardiac defects | 22% |
Epidemiology
The incidence of EA with or without TEF is approximately 2 in 10,000 births with a slight male predominance. Whites tend to be affected more often than non-whites. Increased incidence is seen in first pregnancies and advanced maternal age; the rate of chromosomal abnormalities and rate of twin pregnancy is increased from rates in the general population .
The most common anomaly is EA with distal TEF (Type C, 85%–90%), followed by EA alone (Type A, 7%–8%), TEF without EA (Type E, 4%–5%), EA with proximal TEF (Type B, ∼1%), and EA with fistula to both pouches (Type E, ∼1%) . Associated congenital malformations are seen in approximately 50% of the cases; other gastrointestinal malformations are seen in 25% of the cases, and VACTERL association (vertebral/vascular, anorectal, cardiac, tracheoesophageal, radial/renal, limb deformities) occur in 20% of patients who have EA ( Table 3 ) .
| Anomaly | Incidence |
|---|---|
| Cardiovascular | 35% |
| Gastrointestinal | 25% |
| Neurologic | 25% |
| Musculoskeletal | 20% |
| Renal | 50% |
| VACTERL association | 20% |
| Overall incidence | 50% |
Embryogenesis of esophageal atresia
According to the classic theory, aberrant formation of the tracheoesophageal septum results from abnormal fusion of the lateral folds and may result in anomalous connections between the respiratory and digestive tracts. While studying chick embryos, however, Kluth and Fiegel found no evidence of lateral folds during embryogenesis, but depicted a series of cranial and caudal folds in the region of tracheoesophageal separation. According to the chick embryo model, failure of appropriate development of these folds results in EA or tracheoesophageal fistula. Other investigators further demonstrated the role of epithelial proliferation and apoptosis in the development of the tracheoesophageal septum using the Adriamycin rat model .
Significant association of EA/TEF with other congenital anomalies implies a genetic link and has encouraged a search for possible responsible genes or chromosomal abnormalities. In various animal models tracheoesophageal malformations have been associated with mutations in the retinoic acid receptor ( RAR ), retinoid X receptor ( RXR ), sonic hedgehog ( shh ), thyroid transcription factor 1 ( TTF-1 ), Gli family of zinc-finger transcription factors ( Gli1 , Gli2 , Gli3 ), forkhead transcription factor ( Foxf1 ), homeobox c4 ( Hoxc4 ), and T-box transcription factor ( Tbx4 ) . The aberrant development of the digestive and respiratory tracts may be multifactorial and may involve numerous genetic and environmental factors that have yet to be elucidated.
Diagnosis
Prenatal diagnosis can be established by the presence of polyhydramnios, prominent esophageal pouch, and small or absent stomach “bubble” with fluid-filled loops of bowel on ultrasonography, usually in the third trimester of pregnancy. Polyhydramnios results from the inability of amniotic fluid to reach the stomach in pure EA and decreased amniotic fluid ingestion in cases of TEF with or without EA. The positive predictive value of both polyhydramnios and a small or absent stomach is only 56%. Additional finding of the upper neck pouch sign, which is visualized during fetal swallowing, has high specificity for EA but may need a highly experienced operator .
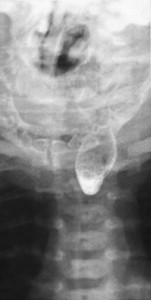
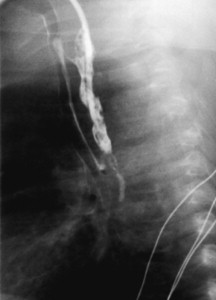
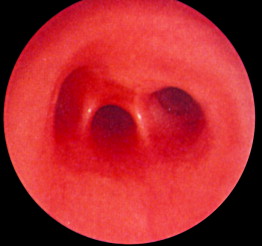
Symptoms suspicious for EA/TEF can be observed shortly after birth and may include: excessive salivation; regurgitation, coughing, and choking following initial attempts to feed the neonate; respiratory distress; cyanosis; or inability to advance a catheter into the stomach. The nasogastric tube may curl in the proximal pouch suggesting atresia ( Fig. 7 ). A distended, air-filled abdomen is suggestive of EA with a distal TEF (Type C) because of the passage of air from the trachea into the gastrointestinal tract. Findings on chest and abdominal radiograph include an air-filled blind pouch with EA, presence of air in the gastrointestinal tract in cases of distal TEF, and pneumonia because of the presence of gastric reflux into the lung from a distal TEF. If there is difficulty in making a diagnosis, a small quantity of nonionic water-soluble contrast or diluted barium can help outline the proximal pouch ( Fig. 8 ). The presence of a proximal air-filled pouch and a gasless abdomen is suggestive of EA without TEF (Type A). Diagnosis of H-type fistula (type E) is usually made using nonionic water-soluble contrast or diluted barium during a radiographic study ( Fig. 9 ). Demonstration of TEF with bronchoscopy can be helpful in diagnosis and operative planning ( Fig. 10 ). The use of CT, virtual bronchoscopy, and MRI has also been reported, although their role in the diagnosis is not yet clear .
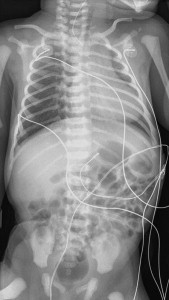
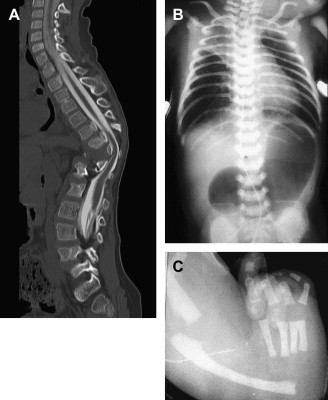
The presence of components of the VACTERL association should increase suspicion for EA. Approximately 50% of patients who have EA or TEF present with additional birth defects. Complete evaluation should include: plain radiographs of the chest, abdomen, pelvis, and spine; ultrasound exams of the spine and kidneys; and echocardiography of the heart and aorta ( Fig. 11 ).
Management
Preoperative
The main goal of preoperative management is to maintain the airway and to prevent lung damage from aspiration of gastric contents. This goal can be achieved by elevating the head of the bed and placing a suction catheter in the proximal pouch to prevent accumulation of saliva and mucus. Preoperative diagnostic studies should be performed expeditiously; echocardiographic evaluation of the heart and aorta are valuable in deciding the operative approach. The right chest is the preferred approach when a normal left-sided aortic arch is demonstrated.
Operative
Thoracotomy
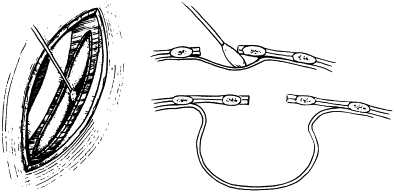
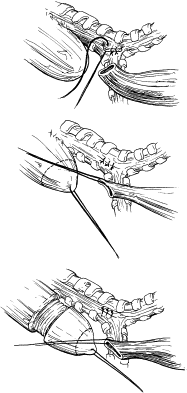
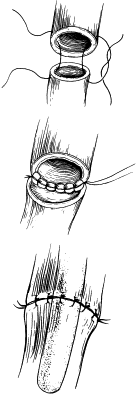
The type of EA/TEF along with the length of the gap determines the timing and approach of the repair. The most common presentation, EA with distal TEF (type C), warrants an attempt at primary repair using the right-sided posterolateral extrapleural approach at the level of the fourth intercostal space ( Fig. 12 ). The proximal pouch is mobilized in the cephalad direction to allow for approximation to the distal segment of the esophagus. The fistula is then carefully dissected to avoid damaging the trachea. The vagus nerve may be visualized as a landmark to identify the distal pouch as it enters the trachea; during this dissection care should be taken to avoid damage to its esophageal branches. The fistula is circumferentially mobilized and incised close to the trachea, leaving a rim of esophageal tissue on the trachea and placing interrupted nonabsorbable sutures to close the fistula and support the resulting weakening in the trachea ( Fig. 13 ). Dissection of the distal esophagus should be limited to avoid damaging its sparse blood supply. The anastomosis of the proximal esophageal pouch to the distal segment is usually achieved end-to-end using a set of two opposing traction sutures to achieve approximation of the two segments. The next step is placement of interrupted fine absorbable sutures through full thickness of the esophagus beginning at the posterior wall with the knots tied inside the lumen. An 8 French tube is passed down the esophagus with the end distal to the site of anastomosis, usually into the stomach, and interrupted sutures are placed in the anterior wall with extraluminal knots ( Fig. 14 ). If the surgeon observes high anastomotic tension, the Haight two-layer telescoping anastomosis may achieve a better operative outcome. Another approach may be an end-to-side anastomosis or the performance of circular myotomies to gain needed esophageal length. Once the anastomosis is complete, a 10 or 12 French chest tube is usually placed in the area of the repair and secured to the chest wall. Some surgeons elect to place a transanastomotic feeding tube to encourage early enteral feeding. The chest wall is closed in layers with appropriate suture materials .
Thoracoscopy
Advances in pediatric endoscopic surgery have resulted in the application of thoracoscopy to the management of EA. The first report of thoracoscopic repair was published in 1999 by Lobe and colleagues . The advantages of using a minimally invasive approach include decreased morbidity associated with thoracotomy (respiratory compromise, postthoracotomy pain, postoperative recovery time, scoliosis, shoulder girdle weakness, chest wall asymmetry) and improved visualization with a magnifying camera. Disadvantages to performing the repair thoracoscopically include difficulty maneuvering instruments in the small working space and inability to place more than one suture at a time, causing increased tension and increased risk for suture material tearing through the tissue.
Prerequisites to a successful thoracoscopic repair include stable condition of the infant, a surgeon skilled in minimally invasive surgery, and adequate support from the anesthesia team. Absolute contraindications to endoscopic approach include severe hemodynamic instability, significant prematurity, or weight less than 1500 g. Relative contraindications include severe congenital cardiac defects, birth weight 1500 to 2000 g, or significant abdominal distention because of a proximal bowel atresia .
Operative management for thoracoscopic repair includes general anesthesia with endotracheal intubation, preferably of the left mainstem bronchus. The infant is placed in the prone position with the right side elevated to a 45° angle. Proper port placement is essential to obtain good visualization of the thoracic cavity and to allow for necessary instrument manipulation. The first 3- to 5-mm cannula is placed in the posterior axillary line at the level of fifth intercostal space, carbon dioxide is insufflated into the thoracic cavity, and a 30° camera is advanced through the port. One 5-mm port is placed in the midaxillary line one to two intercostal spaces above and one 3-mm port is placed in the midaxillary line one to two intercostal spaces below the laparoscope port; both are used for instruments, which should approximate at about a 90° angle. A fourth port is occasionally needed to achieve good lung retraction, although it is usually unnecessary. Once the fistula and the proximal pouch are identified, the surgeon should proceed with the dissection, division of the fistula, and anastomosis of the proximal and distal segments in the same basic steps as those described for thoracotomy .
Repair of long-gap atresia
Long-gap EA (usually type A) is defined as a gap of approximately 2.5 cm with the two segments not readily approximated without considerable tension. The traditional approach to the management of long-gap EA/TEF is to perform an initial feeding gastrostomy and to wait several weeks for spontaneous esophageal growth. Although performing the feeding gastrostomy, it is important to try to place the stoma closer to the lesser curvature to preserve the vascular supply of the greater curvature in case a reversed gastric tube is needed for esophageal replacement. Delayed primary repair may be accomplished once the segments are of sufficient length. Howard and Myers pioneered the use of daily bougienage of the proximal pouch to encourage its growth. Placing bougies or contrast in proximal and distal pouches (through the gastrostomy) allows for the gap to be periodically measured with fluoroscopic imaging ( Fig. 15 ). The application of an electromagnetic field to metal devices in the two respective pouches to promote growth and allow the gap to narrow has also been described , as has application of hydrostatic pressure to induce growth in the distal pouch . Other techniques to elongate the esophageal segments included the use of proximal and distal circular myotomies and growth promotion using magnets, stringed beads, guide wires, and steel bars .
John Foker describes an innovative technique for assessment and repair of long-gap EA with or without TEF, which begins with dissection of the proximal and distal pouches and placement of four traction sutures on each segment without entering the lumen ( Fig. 16 ). Primary repair is usually achievable if the segments are easily approximated, whereas if the surgeon perceives a significant amount of tension a multistaged operative approach may be necessary. Such an approach involves placing internal traction sutures from the upper and lower pouches under tension in the prevertebral fascia below and above the thoracotomy incision, respectively; in ultra long-gap these traction sutures are brought out through the intercostal spaces and tied over silastic buttons on the posterior chest wall ( Fig. 17 ). Consequently, tension can be increased daily to promote rapid growth of the two segments. Clips may be placed on the ends of the two segments so that the advancement may be followed by plain film radiography. A second thoracotomy is then performed to achieve a primary end-to-end anastomosis. In Foker’s series , adequate growth of the proximal and distal pouches occurred in 8 to 18 days to allow for primary repair.
Kimura and colleagues developed a technique of multistaged extrathoracic elongation of the proximal esophageal pouch that subsequently permits approximation and anastomosis of the two segments. Initial cervical approach involves freeing the proximal esophageal segment and creating an esophagostomy on the anterior chest wall through which “sham” oral infant feeding is administered and collected in an ostomy bag; nutrition is supplied by a gastrostomy tube until the repair is complete. Two months following the initial operation the esophagostomy is revised and placed lower on the chest wall. Once the proximal and distal pouches are approximated, a definitive end-to-end anastomosis is performed by way of a thoracotomy.
Although added complications of a multistage repair of long-gap atresia exist, the benefits of attaining repair of the native esophagus clearly decrease life-long morbidities for the patient. Complications of a multistaged repair include adhesions, anastomotic leaks, development of strictures (usually successfully managed using dilation), recurrence of fistulas, disorders of motility, and gastroesophageal reflux. Nevertheless, anastomosis of the native esophageal segments is a better option in the long term for patients who have long-gap atresia as compared with esophageal replacement .
Esophageal replacement
Esophageal replacement is necessary for patients in whom repair of the native esophagus is not feasible. Various techniques for replacement have been described using the stomach, small intestine, and colon. Gastric transposition to reestablish continuity of the upper gastrointestinal tract has been found to be effective in a large series of patients . Another technique, first described by Schärli, uses the lesser curvature of the stomach to create a 3- to 4-cm tube for replacement of necessary esophageal length . Similar techniques may be used to create a reversed or isoperistaltic gastric tube using the greater curvature of the stomach . Likewise, the uses of jejunal, ileal, or colonic segments for esophageal replacement have been described. Complications of these operations include dysmotility, dysphagia, difficulty feeding, gastroesophageal reflux, stricture, dilation of the nonnative segment, fistula formation, and compromised pulmonary function. Although the survival rate for these patients is good (∼95%), associated morbidities often require additional procedures or operations .
H-type tracheoesophageal fistula
Diagnosis of congenital H-type tracheoesophageal fistula (type E) may be challenging. Common presenting symptoms suspicious for H-type tracheoesophageal fistula (type E) include cough and choking during feeding, recurrent pulmonary infections, and aerophagia. Anatomically, the fistula courses in an oblique fashion from the posterior aspect of the trachea to the more caudal anterior aspect of the esophagus. If H-type TEF is suspected, a contrast esophagogram is usually diagnostic (see Fig. 9 ). Bronchoscopy at the time of surgery, confirms the anatomic location of the fistula, which is important in operative planning and allows placement of a catheter in the fistula tract. Generally if the fistula is at the level of the second thoracic vertebra (T2) or higher (70% of H-type TEF), repair should proceed through a cervical incision, whereas fistulae below T2 require a thoracic approach. Thoracoscopic and endoscopic repairs have also been described .
For high fistulae, a right cervical incision is made and the fistula identified with the assistance of intraoperative bronchoscopic catheter placement. The fistula is then approached by dissection within the tracheoesophageal groove. The recurrent laryngeal nerve should be identified and preserved, the fistula is ligated and divided, and the esophageal and tracheal openings are closed using single-layer interrupted sutures. A local muscle flap may be transposed between the repaired trachea and esophagus. A nasogastric tube is placed to provide enteral nutrition and is usually removed around the fifth postoperative day. The success of operative treatment is documented by contrast esophagogram on about postoperative day five, after which oral feeds are resumed .
Postoperative
In an uncomplicated repair without anastomotic tension, prompt extubation is desirable if the cardiopulmonary status is stable. If excessive tension is noted, some surgeons prefer keeping the infant intubated and on neuromuscular blockade, with the neck in flexion. The head of the bed should be elevated to minimize the amount of gastroesophageal reflux. If a transanastomotic feeding tube has been placed, early enteral feeding may be instituted with return of gastrointestinal motility. If a feeding tube has not been placed, a contrast esophagogram should be performed around the fifth postoperative day and if no leak is noted oral feeds may be initiated and the retropleural drain removed. If the contrast esophagogram demonstrates a leak, or if saliva is noted in the draining retropleural fluid, feeding is delayed and drain output is recorded to monitor the progression of the leak. Most small leaks can be managed conservatively and resolve spontaneously without need for reoperation.
Outcomes
Immediate postoperative complications and long-term outcomes for repair of EA with distal TEF (type C) are tabulated ( Table 4 ) .



