Microtia and congenital aural atresia (CAA) are congenital anomalies that are so common that every otolaryngologist should be familiar with the initial evaluation and care of the patient. When one ear hears normally, speech and language development should be normal. The gross and fine motor development of the baby or child is not expected to be affected in isolated cases of microtia and CAA. Current technologies allow for reconstruction or habilitation of the microtic ear when the child is several years of age. The hope is that tissue engineering can eliminate donor site morbidity. Temporary prosthetic ears will remain an option. Aural atresia work continues to be very dependent on the patient anatomy and the need or desire for better hearing in the affected ear.
Microtia is the abnormal development of the external ear that results in a malformed auricle. The deformity that results can range from mild distortion of the anatomic landmarks to the complete absence of the ear ( Figs. 1–3 ). Congenital aural atresia (CAA), which is commonly associated with microtia, is the failure of the development of the external auditory canal (EAC).
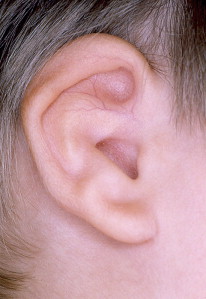
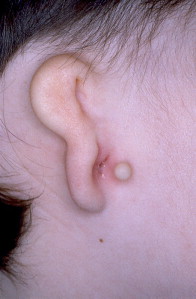
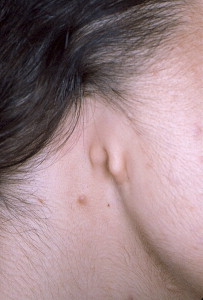
Microtia and CAA are relatively rare deformities; still, an otolaryngologist can expect to be called on to evaluate this defect several times in the average career. Although not every otolaryngologist will perform the complex surgical reconstruction of these malformations, one should be familiar with the etiology, anatomy, medical management, and nonsurgical options available for microtia and CAA. The goal of this article is to provide an overview of these areas, and to give an update of current trends and future directions.
These deformities do not exist independent of other malformations of the auditory system. A discussion of middle and inner ear abnormalities is covered elsewhere in this issue.
Epidemiology and etiology
A number of causes have been implicated in the genesis of microtia, including teratogens (such as vitamin A) , vascular insults, and genetic aberrations. Isolated microtia results when the branchial arches develop abnormally. When part of a syndrome, microtia/atresia may be a result of a single gene deletion, or the result of embryopathic development, such as in Goldenhar syndrome. Some auricular deformities are a result of multifactorial insults to the fetus . Microtia and CAA occur in approximately 1 in 10,000 to 20,000 births. Both occur more often unilaterally, with a predilection for the right side. Men are affected more than women, at a 2.5:1 ratio. Microtia is commonly associated with CAA, and the degree of auricular malformation usually correlates with the degree of middle ear deformity. However, the incidence of inner ear abnormalities is relatively low in patients who have CAA . Microtia is also associated with other anomalies approximately 50% of the time, especially the facioauriculovertebral syndromes. Microtia is seen in higher numbers in Japanese, Hispanic, and Native American populations (particularly the Eskimo and Navajo) . Women with four or more pregnancies are at increased risk for having a child with microtia, especially its most severe form, anotia . CAA is more often bony than membranous.
Embryology
During the sixth week of gestation, the external ear begins to develop around the dorsal end of the first branchial cleft. On either side of the cleft lies the first (mandibular) and second (hyoid) branchial arches. The auricle arises from these arches as six small buds of mesenchyme, known traditionally as the six hillocks of His. The mandibular arch gives rise to hillocks 1 through 3, and the hyoid arch gives rise to hillocks 4 through 6 . Traditionally, it is thought that the derivation of specific auricle components comes from certain hillocks (ie, hillock 1 becomes the tragus, hillocks 2 and 3 form the helix, hillocks 4 and 5 form the antihelix, and hillock 6 forms the lobule). Other theories suggest that the hyoid arch contributes approximately 85% of the auricle, and that most of the central ear is formed from hillocks 4 and 5, whereas the tragus is formed from hillocks 1 through 3. The lobule is the last component of the external ear to develop. The concha is derived from the ectoderm forming the EAC . The auricle begins in the anterior neck region, then migrates dorsally and cephalad as the mandible develops during gestational weeks 8 through 12, and lies in its relative adult location by gestational week 20 .
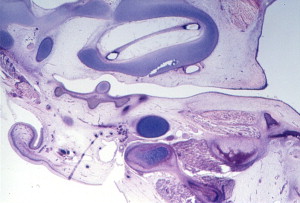
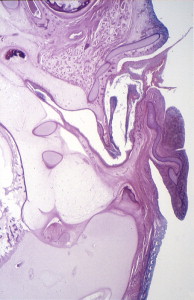
During the first and second months’ gestation, the external auditory meatus develops from the first branchial groove between the mandibular and hyoid arches. At 4 to 5 weeks’ gestation, a solid epithelial nest forms at the meatus, and contacts the endoderm of the first pharyngeal pouch. Mesoderm interrupts the contact between the endoderm and ectoderm. At 8 weeks, the cavum conchae invaginates, forming the primary meatus, which becomes surrounded by cartilage and eventually becomes the fibrocartilaginous portion of the EAC. This groove deepens and grows toward the tympanic cavity and comes into contact with the epithelium of the first pharyngeal pouch. A solid epidermal plug extends from the primary meatus to the primitive tympanic cavity to form the meatal plate ( Fig. 4 ). This plug of tissue recanalizes during the twenty-first week, forming the primitive EAC, and by the twenty-eighth week, only the most medial ectoderm remains, forming the superficial layer of the tympanic membrane ( Fig. 5 ). The ectoderm lines the bony portion of the EAC .
Embryology
During the sixth week of gestation, the external ear begins to develop around the dorsal end of the first branchial cleft. On either side of the cleft lies the first (mandibular) and second (hyoid) branchial arches. The auricle arises from these arches as six small buds of mesenchyme, known traditionally as the six hillocks of His. The mandibular arch gives rise to hillocks 1 through 3, and the hyoid arch gives rise to hillocks 4 through 6 . Traditionally, it is thought that the derivation of specific auricle components comes from certain hillocks (ie, hillock 1 becomes the tragus, hillocks 2 and 3 form the helix, hillocks 4 and 5 form the antihelix, and hillock 6 forms the lobule). Other theories suggest that the hyoid arch contributes approximately 85% of the auricle, and that most of the central ear is formed from hillocks 4 and 5, whereas the tragus is formed from hillocks 1 through 3. The lobule is the last component of the external ear to develop. The concha is derived from the ectoderm forming the EAC . The auricle begins in the anterior neck region, then migrates dorsally and cephalad as the mandible develops during gestational weeks 8 through 12, and lies in its relative adult location by gestational week 20 .
During the first and second months’ gestation, the external auditory meatus develops from the first branchial groove between the mandibular and hyoid arches. At 4 to 5 weeks’ gestation, a solid epithelial nest forms at the meatus, and contacts the endoderm of the first pharyngeal pouch. Mesoderm interrupts the contact between the endoderm and ectoderm. At 8 weeks, the cavum conchae invaginates, forming the primary meatus, which becomes surrounded by cartilage and eventually becomes the fibrocartilaginous portion of the EAC. This groove deepens and grows toward the tympanic cavity and comes into contact with the epithelium of the first pharyngeal pouch. A solid epidermal plug extends from the primary meatus to the primitive tympanic cavity to form the meatal plate ( Fig. 4 ). This plug of tissue recanalizes during the twenty-first week, forming the primitive EAC, and by the twenty-eighth week, only the most medial ectoderm remains, forming the superficial layer of the tympanic membrane ( Fig. 5 ). The ectoderm lines the bony portion of the EAC .
Anatomy
The fully formed auricle is a complex, three-dimensional structure with three portions, the helical–antihelical complex, the conchal complex, and the lobule. The elastic cartilage of the auricle is flexible but strong, and is covered loosely with fibrofatty tissue over the margin of the helix and the lobule; adherent skin covers the rest of the cartilaginous framework ( Fig. 6 ) .
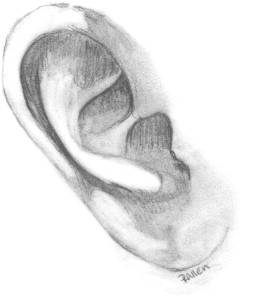
Ears reach mature size earlier in females than males, at 13 and 15 years, respectively. Normal ear height is between 5.5 and 6.5 cm. The helical rim protrudes from the mastoid surface around 1.5 to 2.0 cm, creating an angle of 15 to 20 degrees . The projection increases from superior to inferior, with the helical rim positioned 10 to 12 mm from the skull, and the lower ear 20 to 22 mm from the skull. The superior edge of the auricle should be in line with the lateral edge of the eyebrow or the level of the upper eyelid . The ear is inclined posteriorly anywhere from 5 to about 30 degrees along its vertical axis. Classic teaching states that the angle is parallel to the dorsum of the nose, which is not necessarily true, although the two angles are usually within 15 degrees of each other . The nose is the more variable angle. With a high nasion, the nose is more upright, but if the nasion is lower, the radix is flatter and may not be parallel with the axis of the ear ( Fig. 7 ).
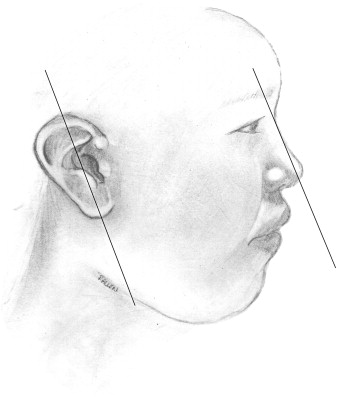
The primary blood supply to the auricle is by way of the superficial temporal artery and the posterior auricular artery. Multiple communications between the two arteries allow one of the vessels to supply the ear if necessary. Venous drainage of the ear is by way of the posterior auricular veins that drain into the superficial temporal, external jugular, and retromandibular veins. The great auricular nerve (C3) supplies most of sensation to the external ear, along with small contributions from the lesser occipital nerve (C2,3), auriculotemporal nerve (V3), and cranial nerves VII and X. The ear is attached to the cranium by way of skin, cartilage, and the extrinsic auricular muscles. These extrinsic muscles include the anterior, superior, and posterior suspensory muscles. Intrinsic muscles are present, but play no role in the support or function of the ear .
The EAC is composed of a lateral cartilaginous (one third) and medial bony (two thirds) canal. The cartilaginous portion is covered with loose skin containing sebaceous glands, ceruminous glands, and hair follicles. The skin overlying the bony canal is thin and tightly adherent. The EAC is curved in a superior and anterior direction .
Initial evaluation
During the initial physical examination, close attention should be paid to the mandible, oral cavity, cervical spine, and eyes, to rule out associated anomalies. Evaluation of the malformed ear should include the quality of the skin, the hairline, and the position of the remnant ear. The integrity of the facial nerve on the malformed side should be assessed because microtia and facial nerve dysfunction are often associated .
Patients who have unilateral microtia and CAA usually have normal hearing in the contralateral ear . Hearing status, however, is the first thing that should be evaluated in the patient lacking other major developmental anomalies. Sensorineural, conductive, and mixed losses can be seen in the microtic/atretic ear Conductive hearing loss represents 80% to 90% of the loss in these patients; however, approximately 10% to 15% will have a sensorineural hearing loss (SNHL) that must be addressed . Patients who have Goldenhar syndrome may have hearing loss in the nonmicrotic ear that is conductive or sensorineural . The threshold of conductive loss on the affected side is expected to be between 40 and 60 dB . In most states, universal newborn hearing screening is mandatory. If the nonmicrotic ear passes the newborn hearing screen, additional testing can be delayed until age 6 to 7 months because hearing is adequate for the acquisition of speech and language. Any hearing concerns before this time require that diagnostic auditory brainstem response testing be performed to be sure at least one ear has normal hearing. In patients who have one malformed ear, careful attention is paid to the normal ear. Any threat to a normal hearing ear should be dealt with promptly. Middle ear effusions should be treated aggressively .
In cases of bilateral atresia, bilateral bone conduction hearing aids should be placed . Traditionally, hearing amplification is not required in unilateral atresia. However, it is now well-established that binaural hearing is superior to monaural in terms of sound localization and speech perception. Children with a unilateral hearing loss are at a greater risk of developing delayed language development and attention deficit disorders, and of performing poorly in school. The plasticity of the developing auditory system benefits from binaural stimulation, which has led to a push toward improving hearing in patients who have unilateral deficits as well, by way of a surgical correction of CAA (after microtia repair) or by placement of a hearing amplification device . Current Food and Drug Administration indications for bone-anchored amplification include age more than 5 years. Typically, microtia repair is performed no earlier than age 6 years, so that CAA drill-out is not before age 7 years. It is unknown if opening the ear at this later time allows for binaural brain development, or if a sensitive period in development has already passed.
CT can be performed to assess middle and inner ear anomalies. Some recommend obtaining this information as early as possible, when the child is several months old, because cholesteatoma is estimated to be present in 4% to 7% of atretic ears and should be ruled out. However, a congenital cholesteatoma often requires several years to grow enough to be evident or problematic, so the CT scan may be delayed until an age where sedation is not required, usually at age 4. If obtained in the newborn period, CT will need to be repeated later if a drill-out procedure for CAA is contemplated, because the temporal bone matures and changes in the first years of life. When obtained at the approximate age of 4, only one CT scan is required. The presence of cholesteatoma or otitis media that is refractory to antibiotics is an indication for surgery, even if a good hearing result is not anticipated .
Congenital ear malformations are not detected readily before birth, and may cause psychosocial trauma to the parents and family . Counseling the family members is an important part of the management of these conditions. The expectations of the parents and the reality of the problem may not coincide. It is important to communicate to the family that although microtia reconstruction will improve the appearance of the auricle, the ear may never look normal. Likewise, operations to improve hearing loss caused by CAA rarely achieve normal hearing. It is important that children with these malformations are evaluated early to rule out associated problems, evaluate hearing status, and develop a plan of management.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


